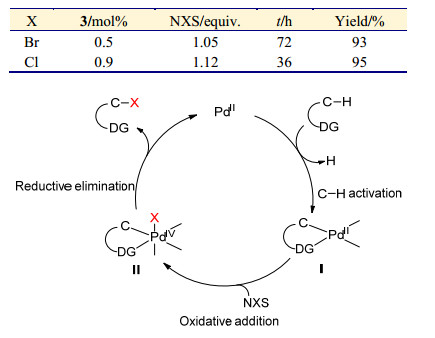
 图式1
钯催化邻位C—H键卤化反应可能机理
Scheme1.
Proposed mechanism for Pd-catalyzed ortho-halogenation
图式1
钯催化邻位C—H键卤化反应可能机理
Scheme1.
Proposed mechanism for Pd-catalyzed ortho-halogenation
引用本文:
骆钧飞, 徐星, 赵延超, 梁洪泽. N-卤代琥珀酰亚胺参与的过渡金属催化芳基C—H键卤化反应研究进展[J]. 有机化学,
2017, 37(11): 2873-2882.
doi:
10.6023/cjoc201705018

Citation: Luo Junfei, Xu Xing, Zhao Yanchao, Liang Hongze. Advance of N-Halosuccinimides in Transition-Metal-Catalyzed Halogenation of Aromatic C-H Bonds[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2873-2882. doi: 10.6023/cjoc201705018
Citation: Luo Junfei, Xu Xing, Zhao Yanchao, Liang Hongze. Advance of N-Halosuccinimides in Transition-Metal-Catalyzed Halogenation of Aromatic C-H Bonds[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2873-2882. doi: 10.6023/cjoc201705018
N-卤代琥珀酰亚胺参与的过渡金属催化芳基C—H键卤化反应研究进展
English
Advance of N-Halosuccinimides in Transition-Metal-Catalyzed Halogenation of Aromatic C-H Bonds
Abstract:
Carbon-halogen (C-X) bonds are amongst the most fundamental groups in organic synthesis, which are frequently and widely employed in the synthesis of numerous organic products. The generation of C-X bond, therefore, constitutes an issue of high interest. Herein, the research advances on the transition-metal-catalyzed C-X (X=Cl, Br, I) bond formation via direct C-H bond of aromatic compounds transformation by the use of N-halosuccinimide (NXS) is reviewed.
-
Key words:
- N-halosuccinimide
- / C-X bond
- / C-H bond activation
- / transition-metal catalysis
-
C—X键(X=Cl, Br, I)广泛存在于生物活性分子、药物分子以及功能材料分子中[1].另外, 由于C—X键多样的反应活性, 有机卤化物是有机合成中一类非常重要的中间体[2].因此, C—X键的构建方法研究受到了有机合成化学家的广泛关注.卤代芳香环化合物在有机合成中是一类非常重要的反应底物, 比如在经典的合成反应中, 通过卤代芳香环可以制备有机锂试剂[3]、格氏试剂[4]以及苯炔中间体[5], 并且其通过亲核取代反应可以得到多样性官能团取代的芳香环化合物[6].在近代有机合成中, 卤代芳香环化合物被广泛应用于贵金属催化的交叉偶联反应中, 高效构建多种C—C键[7]、C—N键和C—O键[8].因此, 将芳香环的C—H键选择性转化为C—X键在有机合成中具有极其重要的意义.
最常见的芳香环化合物C—H键卤代方法是通过卤素气体、过氧化物/HX、过氧化物/MX或者高价碘试剂/MX (M=Li, Na, K或TMS)进行亲电芳香取代反应(SEAr)[9].尽管这些传统合成方法已经得到了广泛的运用, 但是依然存在一些不足, 例如: (1)底物范围受到活泼芳香环的限制; (2)通常伴有各种各样的副反应, 如苄基卤化和过度卤化等; (3)芳香族卤代模式具有其自身局限性; (4)产物易产生位置异构, 导致产率降低, 分离难度增大[10].另一重要的芳香环卤代的方法是通过强碱丁基锂试剂进行锂化反应得到有机锂化合物, 有机锂化合物通过卤素猝灭生成卤代芳香环化合物[11].该方法通常用于各种复杂分子的反应当中.然而, 该反应也受到各种限制, 强碱丁基锂的使用需要严格控制无水无氧操作, 反应的官能团耐受性低.因此, 发展新型的高选择性和广泛官能团兼容性的芳香环C—H卤化反应具有重要的意义.关于这一反应已有不少综述分别从不同角度做了总结, 比如Iskra等[12]探讨了氧气和双氧水作为绿色氧化剂实现氧化卤化反应的研究.刘云云等[13]基于铜催化的卤化反应进行了阐述.史炳锋等[14]概述了不活泼C—H键[包括C(sp2)—H键和C(sp3)—H键]的卤化反应研究. Sutherland等[15]则围绕过渡金属催化芳香环的碘化进行了讨论.
N-卤代琥珀酰亚胺(NXS)由于N—X键的高极化性, 在不同的条件下, 可通过不同的裂解方式选择性形成卤素自由基、卤素阴离子、卤素阳离子等, 从而分别通过自由基反应、亲电取代反应以及亲核取代反应构建C—X键, 因此其被广泛地应用于卤化反应中[16].相比其它卤代试剂, NXS具有独特的优点, 例如: (1)与卤素气体卤素源相比, NXS的使用更加方便; (2)与高价碘试剂/MX卤素源相比, NXS制备简单, 且化学性质稳定; (3)与过氧化物/HX卤素源相比, NXS的使用更加安全可靠; (4) NXS参与的卤化反应条件温和.然而, 使用NXS进行卤化反应时其反应活性往往在较为活泼的反应位点(比如烯丙基或者苄基位置[17])进行.卤代芳香化合物作为重要的有机分子片段以及合成中间体, 使用N-卤代琥珀酰亚胺来构建芳香环的C—X键时, 面临着区域选择性差的问题.伴随着导向过渡金属催化反应的发展, NXS参与的过渡金属催化下选择性C—H键卤化反应研究得到了快速的发展.本文根据过渡金属催化剂类型的不同, 对近年来N-卤代琥珀酰亚胺作为卤源的芳香环C—H键选择性卤化的方法进行了综述.
1 NXS参与的钯金属催化的芳香环卤化反应
近年来, 过渡金属催化的C—H键活化备受关注, 并已成为在有机化合物中引入特定官能团的高效策略, 从而使过渡金属催化的卤化反应也逐渐成为构建C—X键的强有力工具之一.与传统的C—X键构建方法相比, 过渡金属催化的卤化不仅具有非常高的选择性, 而且可以控制单卤代.钯金属催化C—H键官能团化是目前为止发展最为成熟、研究最多的C—H键官能团化反应.
早在1970年Fahey小组就报道了钯催化偶氮苯与氯气的卤化反应, 但是直到21世纪初才由Sanford课题组[18]报道了NXS参与的钯金属催化的卤化反应(Eq. 1).该方法通过吡啶导向基团与二价钯金属(PdⅡ)催化剂配位, 进而导向活化吡啶邻位的C—H键, 形成环钯(PdⅡ)中间体Ⅰ.中间体Ⅰ在氧化剂的作用下通过氧化加成得到四价钯中间体Ⅱ.最后, 四价钯中间体Ⅱ通过分子内直接还原消除得到卤代产物以及重新生成二价钯金属催化剂(Scheme 1).相比较于氯气作为卤素源的钯金属催化反应, NXS参与的钯催化卤化反应条件更加的温和, 且选择性更高.在随后的时间里, NXS参与的钯催化的高选择性C—H键卤化方法得到迅速的发展.
图式1
钯催化邻位C—H键卤化反应可能机理
Scheme1.
Proposed mechanism for Pd-catalyzed ortho-halogenation
2006年, Sanford课题组[19]又进一步发展了该类型的反应(Eq. 2).大量的导向基比如吡啶、肟醚、异喹啉、异噁唑啉、甲基四氮唑、吡唑、二亚胺、酰胺等都被证实可以与钯金属发生螯合, 从而导向芳香环邻位的C— H键卤化反应.值得注意的是, 对于15为导向基的底物, 在有Pd催化的条件下, 有导向基15的底物与N-溴代琥珀酰亚胺(NBS)、N-碘代琥珀酰亚胺(NIS)试剂反应时, 主要产物均为对位取代, 而与N-氯代琥珀酰亚胺(NCS)试剂反应的氯化产物则主要发生邻位取代.另外, 有Pd催化的15导向基底物溴代产物区域位置异构体的产量比例非常高, 其对位/邻位取代产物比例达到3:1~4:1.其他类型导向基则没有出现上述现象, 都只发生邻位取代.
随后, Sanford课题组[20]对该钯催化导向的芳香环邻位C—H键卤化的作用机理做了深入的研究.该课题组通过对2-芳环吡啶分子内的动力学同位素效应(Kine-tic Isotope Effect, KIE)研究, 指出导向基配位二价钯金属活化邻位C—H键形成环钯中间体是该反应的决速步骤之后对钯催化剂, 反应底物以及NCS试剂做了动力学级数的研究发现金属钯符合一级反应, 而反应底物以及NCS动力学级数为零.作者通过以上数据推测, 首先二价金属钯催化剂PdCl2与过量的底物16配位形成单体金属钯分子17, 底物则作为金属钯的配体.在这之后, 环钯中间体可以通过以下两条途径得到: (a)单体金属钯分子17可以通过五元配位过渡态18得到环钯中间体20; (b)单体金属钯分子通过可逆的配体交换得到阳离子金属化合物19, C—H活化得到环钯中间体20 (Scheme 2).两条途径都符合钯的一级反应动力学以及底物零级反应动力学.作者认为反应经历第二条途径的可能性更大, 因为它能更好解释Hammett ρ值为-0.43的实验事实.
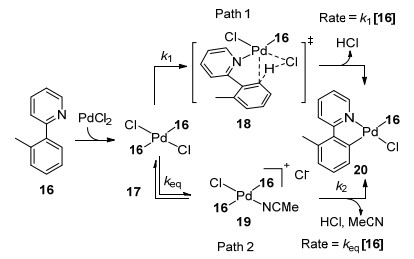 图式2
环钯中间体形成可能机理
Scheme2.
Proposed mechanism for the formation of cyclopalladated complexes
图式2
环钯中间体形成可能机理
Scheme2.
Proposed mechanism for the formation of cyclopalladated complexes
继Sanford课题组之后, 以NXS为卤源的过渡金属催化芳香环C—H键卤代反应不断被探索. 2011年, Bedford课题组[21]报道了温和条件下钯催化酰基苯胺的邻位卤化(Cl, Br)反应, 其优点在于反应常温下就可以进行, 且无需无水无氧操作, 同时反应时间也大幅度缩短到1~4 h.值得指出的是, 其首次在反应条件中加入了对甲苯磺酸(PTSA) (Eq. 3). PTSA可能的作用是使NXS的羰基质子化使其成为一个更有效的X+源.相比较于NXS, 质子化的NXS(也有可能原位生成HOX)更有利于氧化加成环钯中间体, 最后还原消除得到卤代产物.该方法具有较好的普适性, 下面将列举一些基于该机理但添加的酸有所区别的报道.
PTSA促进此反应进行的可能机理的探究过程值得一提.基于酰基苯胺环钯中间体实现C—H键的活化被广泛接受[22], 但是作者通过实验发现使用基于酰基苯胺环钯中间体进行卤化反应时存在至少20 min的反应诱导期, 而在标准条件下不存在这个诱导期.随后作者通过大量实验发现一个基于PTSA邻位钯化的化合物23.使用化合物23进行卤化反应结果与标准条件下的卤化反应结果一致.此外, 化合物23在与类似标准反应的条件下发生自身还原得到PdⅠ-PdⅡ四聚体24, 这些实验事实仍然不能确定该反应是否经历三价钯中间体(PdⅢ)亦或四价钯中间体(PdⅣ).
在这之后, 在温和条件下, 基于有机强酸促进的NXS参与的钯金属催化芳香环C—H键的卤化的方法被相继报道. 2013年, Punniyamurthy课题组[23]使用三氟甲磺酸(CF3SO3H)作为添加剂, 在较温和的条件下, 以NXS为卤源, 醋酸钯为催化剂, 实现了氨基四氮唑为导向基的邻位C—H键卤化(Eq. 4), 该方法区域选择性好, 目标产物产率高, 底物兼容性良好.
同年孙萌课题组[24]报道了相同机理下NXS为卤源、Pd(OAc)2催化的氧化偶氮苯类的邻位C—H卤代(Eq. 5), 该策略反应条件温和, 催化效率高.值得注意的是, 该方法在进行溴代时, 使用二氯甲烷(DCM)作为溶剂, 并添加了TsOH可以大幅度提高反应产率, 达到65%~98%, 且反应温度相对较低(40 ℃).但是在进行氯代、碘代时, 以醋酸(AcOH)直接作为溶剂并未添加其他强酸得到更好的产率, 与溴化条件相比较, 反应所需温度相对较高(Cl: 100 ℃, I: 60 ℃).随后, 孙萌课题组[25]在类似条件下实现了2-吡啶亚砜邻位的C—H卤化反应. Fabis课题组[26]使用了大量的三氟乙酸(TFA)实现了对甲苯磺酰胺导向基邻位的C—H键卤化反应, 该方法只需要室温下就能进行, 以70%~89%的产率得到目标产物(Eq. 6).
孙培培课题组[27]发展了以氰基作为导向基的芳香环C—H键卤化反应(Eq. 7).该反应体系中同样使用了对甲苯磺酰胺(PTSA), 用于质子化NXS使其成为更有效的X+源.不同于一般的导向基, 钯金属是通过氰基碳和氮之间的π电子配位的.这种环化中间体的弱配位性可能使其具有较高的反应活性.众所周知, 氰基化合物是重要的有机中间体, 氰基官能团可以快速简捷地转化为羧酸、酯、酰胺、胺、醛、四唑和酮等各种官能团.另外C—X键可以快速转化为C—C、C—N和C—O键, 因此这一研究给有机合成带来了更多的便捷.
该方法被应用到天然化合物Paucifloral F和iso-Paucifloral F的合成中.首先1, 3-二甲氧基-5-苯甲腈(32)与NIS在钯金属催化下反应得到氰基邻位碘化产物33, 33与炔烃34经历Sonogashira反应得到前体Paucifloral F (35)和iso-Paucifloral F (36). 35和36甲氧基脱保护成羟基即得到非手性的天然化合物Paucifloral F和iso-Paucifloral F (Scheme 3).
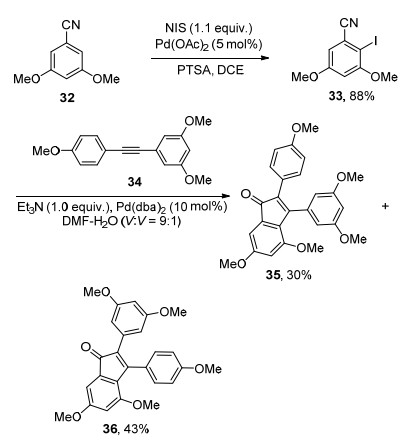 图式3
天然化合物Paucifloral F和iso-Paucifloral F前体的合成
Scheme3.
Synthesis of paucifloral F and iso-paucifloral F precursors
图式3
天然化合物Paucifloral F和iso-Paucifloral F前体的合成
Scheme3.
Synthesis of paucifloral F and iso-paucifloral F precursors
Gevorgyan课题组[28]实现了可脱除导向基二异丙基硅嘧啶邻位的C—H键卤化反应(Eq. 8).值得一提的是, 该导向基可在诱导其邻位C—H键卤化反应后, 进一步诱导其另一个邻位C—H键的烷氧基化, 达到双官能团化的目的.另外导向基二异丙基硅嘧啶可实现多种转化, 比如直接脱除从而得到间位卤代的芳香化合物, 亦可与卤代苯或者炔烃发生偶联反应.
N-杂环卡宾(NHC)可以提供强大的σ-配体和少量的π-反向键合能力来稳定配合物, 因而被广泛用作膦配体的替代物, 但是咪唑类NHC配体化合物有毒、生物降解性差、合成步骤复杂等缺点使其在使用过程中存在一定的影响. 2015年, Purohit课题组[29]则以vitamin B1为配体, 高选择性地实现了吡唑酮邻位的C—H卤化反应(溴化和氯化, Eq. 9).在没有Pd催化剂存在的条件下得到邻位和对位的混合产物.该方法中, 当量银的加入大大提高了产率.作者猜测Ag2O的作用可能是作为氧化剂或者共同催化剂. Ag2O首先氧化底物39邻位的C—H键得到中间体41. Pd金属转移得到环钯中间体42, 然后通过氧化加成和还原消除得到产物40 (Scheme 4).
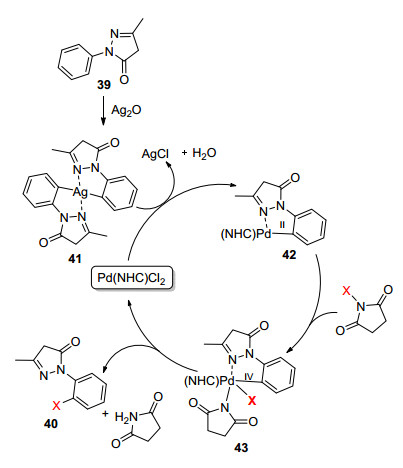 图式4
Pd(NHC)Cl2催化C—H卤化反应
Scheme4.
Pd(NHC)Cl2 catalyzed C—H halogenation
图式4
Pd(NHC)Cl2催化C—H卤化反应
Scheme4.
Pd(NHC)Cl2 catalyzed C—H halogenation
游书力课题组[30]则巧妙地引入含有双配位点的手性配体46, 在Pd金属催化剂存在下, 以NIS为碘源实现了不对称的芳香环C—H键的碘化(Eq. 10).该方法通过动力学拆分的手段, 内消旋体Rac-44在S构型的配体46存在下, 金属Pd催化剂催化得到S构型的碘化产物S-45以及未反应的R构型的底物R-44.该方法是第一个基于Pd(Ⅱ)/Pd(Ⅳ)催化剂循环通过C—H键活化得到轴手性化合物的方法.其ee值也较为理想, 最高可达到94%.可能的机理是氧负离子配位金属钯, 由于手性配体的空间结构, S构型的配体46使得钯选择性活化S构型的44中邻位的C—H键得到环钯化合物47. 47进一步氧化加成得到48, 最终还原消除得到产物(S)-45 (Scheme 5).
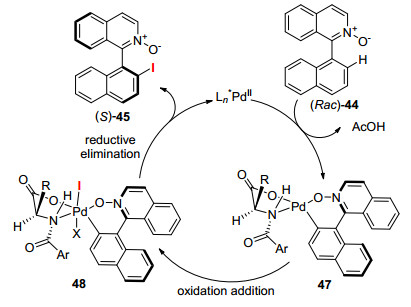 图式5
NIS参与的不对称C—H碘化
Scheme5.
Asymmetric iodination using NIS
图式5
NIS参与的不对称C—H碘化
Scheme5.
Asymmetric iodination using NIS
手性吡啶N-氧化物在不对称的羟醛缩合反应和芳香醛的不对称烯丙基化反应中具有重要的意义[31].而制备手性吡啶N-氧化物往往需要通过钴催化[2+2+2]环三聚反应, 再手性拆分分离得到[32].作者则通过对吡啶N-氧化物不对称碘化得到S-45, 然后将卤化的产物S-45与苯硼酸通过铃木-宫浦交叉偶联反应(Suzuki-Miyaura cross coupling)得到手性吡啶N-氧化物配体49 (Eq. 11), 该配体可应用于芳香醛的不对称烯丙基化反应(Eq. 12).
2 NXS参与的铑金属催化的芳香环卤化反应
钯金属催化的C—H键卤化反应报道层出不穷的同时, 贵金属铑催化的芳香环直接卤化反应方面, 却显得较为欠缺.直到2012年, Glorius课题组[33]首次报道了三价铑(RhⅢ)催化的导向芳基C—H键的氯化和溴化反应(Eq. 13).导向基诸如酰胺、吡啶、酮以及酯等都适应该方法的条件, 导向邻位的C—H键卤化得到高产率的对应产物.该方法兼容多种的官能团包括强给电子基甲氧基以及强吸电子基硝基.动力学同位素效应实验揭示决速步骤是C—H键的活化过程.作者认为第一种反应历程可能类似钯催化的机理, 首先铑催化剂导向活化C—H键形成环铑化合物62, 然后氧化加成以及还原消除得到卤化产物64, 反应循环基于RhⅢ/RhⅤ催化循环.而另一种反应历程可能是环铑化合物62直接与NXS通过SN2反应机理得到产物64 (Scheme 6).
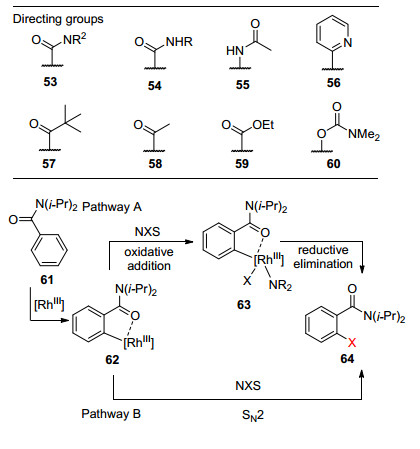 图式6
铑催化的C—H卤化反应
Scheme6.
Rh-catalyzed C—H halogenation
图式6
铑催化的C—H卤化反应
Scheme6.
Rh-catalyzed C—H halogenation
随后, Glorius课题组[34]实现了NIS参与的铑催化富电子杂环的C—H键碘化反应(Scheme 7).该方法适应的杂芳环包括噻吩、呋喃以及吡唑.另一方面, Chang等[35]报道了以NIS以碘源的铑催化喹啉氮氧化物8位的碘代反应(Eq. 14), 该反应条件下分别使用NBS和NCS进行溴化和氯化得到较低的产率. Ding等[36]使用苯并噻唑作为导向基, 在铑催化剂条件下实现邻位的C—H键碘化和溴化反应(Eq. 15).
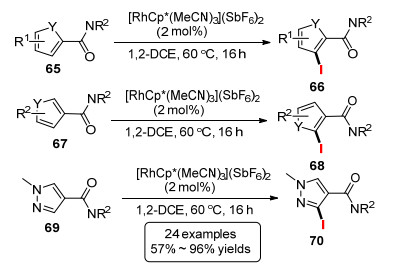 图式7
铑催化杂芳环C—H碘化反应
Scheme7.
Rh-catalyzed heteroaromatics C—H iodination
图式7
铑催化杂芳环C—H碘化反应
Scheme7.
Rh-catalyzed heteroaromatics C—H iodination
Chang课题组[35]将Rh-催化的C—H键卤化方法应用于紫外激发的蓝色荧光锌离子探针zinquin ethyl ester (76)的合成, 首先喹啉氮氧化物的衍生物通过Rh-催化得到8位碘代的产物75, 该产物75通过C-8氨化反应可快速高效地得到76 (Eq. 16).
3 NXS参与的铜催化的芳香环卤化反应
铜作为一种常见的过渡金属, 具有良好的兼容性, 且价格相对低廉, 因而受到广泛关注.另一方面, 鉴于Cu催化的碳杂原子键形成反应的发展取得的显著进展, 受到Pd(Ⅱ)催化剂的启发, 越来越多的注意力转向了Cu催化的C—H官能化反应[37].在C—H键的卤化反应中, 铜试剂, 比如卤化铜试剂往往被用作卤素源, 具有高选择性的特点[38].铜催化的C—H卤化反应较少被报道, 尤其是NXS参与的铜催化C—H卤化反应很少被报道[39].
2013年, Carretero等[40]报道了卤代铜催化吡啶磺酰基保护的苯胺邻位的C—H键溴化和氯化(Eq. 17).作者强调在没有铜催化剂存在下只得到对位的卤代产物.保护基吡啶磺酰基可在镁存在下被脱除从而得到卤代的苯胺.其次, 卤代产物可与炔烃反应生成吲哚.
2015年, 史炳锋课题组[41]报道了高效绿色的铜催化下以NXS为卤源的1-(2-吡啶基)-1-甲基乙胺(PIP)双齿导向基导向的邻位C—H卤化反应(Eq. 18).不同于Carretero等的工作需要一个大气压的氧气作为氧化剂, 该方法只需要在空气条件下就能进行, 最高产率可达90%.并且, 该方法适用于芳香杂环化合物比如噻吩、呋喃和吡啶杂环化合物.值得一提的是, 当量的醋酸锌试剂在反应中具有重要的作用, 作者认为可能是醋酸锌作为路易斯酸活化NXS试剂.卤代产物可在酸性条件下脱除保护基得到邻位卤代的羧酸衍生物.
4 NXS参与的其它金属催化的芳香环卤化反应
而近年来, 相比贵金属催化剂, 较为便宜的金属钌络合物是将惰性C—H键转化为C—C键、C—O键或C—N键过程中的高效催化剂[42], 但是对于C—H键的卤化方面, 适应性仍有待提高.令人惊喜的是, 2014年, Ackermann课题组[43]首次使用苯甲酰胺作为底物, 并使用NXS为卤源, 成功在钌催化下使C—H键转化为C—X键(X=Br, I), 该方法底物适用范围广泛, 官能团兼容性良好(Eq. 19).反应的机理虽未明确, 但是作者在进行动力学同位素效应实验中发现C—H的活化并不是该反应的决速步骤.
最近, Ackermann等[44]突破性地使用了非均相过渡金属Ru催化剂, 实现了碱基间位的C—H溴化反应(Eq. 20).相比较于均相催化剂, 非均相催化剂可以提高过渡金属的重复使用率, 大幅度节省工业成本.该方法是首例使用非均相Ru催化剂催化芳烃间位C—H活化, 反应选择性高, 只得到单一的间位卤化芳环, 更加邻近容易活化的邻位C—H键以及其他具有更低解离能和更强酸性的C—H键没有发生卤化反应.另外, 该方法的普适性良好, 导向基不仅局限于嘌呤, 吡啶和嘧啶亦可在该条件下导向其间位C—H键的卤化反应.作者回收催化剂并重复使用, 发现催化剂在重复使用五次后仍得到超过60%的产物, 再使用七次后得到45%的产物.具体的机理并没有阐述, 但是进一步的动力学实验证实了该反应相对于催化剂Ru是一级动力学反应.使用D2O作为混合溶剂时, 并没有得到显著的H/D交换的产物.
除了钌催化的NXS参与的C—H卤化反应外, 2014年, Glorius课题组[45]首次报道了三价钴(CoⅢ)催化的由吡啶或者酰胺导向基导向的邻位溴化、碘化反应, 并使用NIS和N-溴酞亚胺(NBP)作为卤源(Eq. 21).特戊酸(PivOH)的作用可能是通过质子化NIS或者NBP来增加其亲电性, 或形成具有空位点配位体的活性Co催化剂, 促进反应进行.该方法不仅适用于C—H键的卤化, 还可应用于氰化以及烯丙基化反应.反应可能经历的历程如下:催化剂Cp*CoI2(CO) (85)在AgSbF6和PivOH存在下释放CO配体, 生成活性催化剂86.催化剂86与导向基配位活化邻位C—H键从而形成环钴中间体87, 中间体87的Co原子与NIS中的O原子配位形成88, 最后C—Co键对I—N键进行亲核进攻, 得到卤代产物5和钴化合物89, 89在酸性条件下质子化得到90并重新得到催化剂86 (Scheme 8).
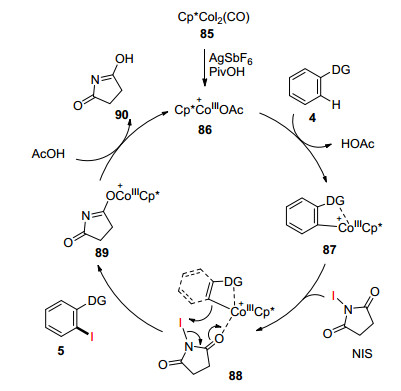 图式8
钴催化的C—H卤化反应
Scheme8.
Co-catalyzed C—H halogenation
图式8
钴催化的C—H卤化反应
Scheme8.
Co-catalyzed C—H halogenation
金属-金属共催化的反应已成为提高有机合成效率的一种有效的策略, 该方法的研究日益受到国内外研究者的关注[46].但是在C—H键的卤化方面金属-金属共催化的鲜有报道, 直到2015年, Ghosh课题组[47]利用非均相Cu-MnO共催化剂, 在可见光的照射下, 实现了芳香族化合物的C—H卤代反应, 该反应使用NXS作为卤化试剂, 可对C—H键进行氯化、溴化以及碘化(Eq. 22).该方法不需要任何的其他的添加剂以及配体, 底物适应范围大, 对于具有富电子和缺电子底物的卤化作用效果均明显, 且选择性良好.此外值得注意的是, 氯代反应可发生在酯基导向的芳香环上, 其产物只要得到间位C—H氯化的产物(Eq. 23).
作者认为该反应的机理可能是通过自由基的机制进行的.首先, 在可见光下, 二价铜和NXS反应生成卤代铜并与底物91配位得到铜化合物95, 95通过单电子转移(SET)得到96, 96中的卤素负离子通过分子内的转移得到中间体97和一价铜, 一价铜可被氧气氧化重新得到二价铜催化剂, 而中间体97通过单电子转移失去一个氢正离子最终得到产物92 (Scheme 9).
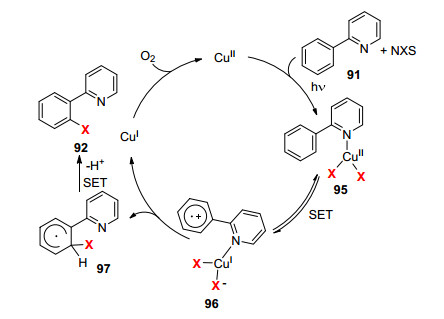 图式9
Cu-MnO催化的C—H卤化反应
Scheme9.
Cu-MnO-catalyzed C—H halogenation
图式9
Cu-MnO催化的C—H卤化反应
Scheme9.
Cu-MnO-catalyzed C—H halogenation
5 结论与展望
综上所述, N-卤代琥珀酰亚胺作为廉价低毒性的卤素试剂, 其参与的过渡金属催化C—H键卤化反应在近几年中取得了快速的发展, 为合成结构多样性的含卤素化合物提供了一种快速有效的策略.迄今为止, 对钯金属催化的芳香环C—H键卤化反应的研究较为成熟, 大量优秀的工作实现了多种底物的C—H键选择性转化为C—X键, 且反应条件相对较为温和.与此同时, 亦有不少文献报道了关于N-卤代琥珀酰亚胺参与的铑催化C—H键的卤化反应.但是相对价格低廉的过渡金属比如铜、钌、钴等催化C—H键的卤化反应的研究很少, 在催化效率、反应条件和底物类型等方面有很大的局限, 因此深入研究发展廉价过渡金属催化的C—H键卤化反应具有很重要的意义.另一方面, 尽管钯金属催化的C—H键卤化反应研究比较成功, 但是都是基于导向基导向的, 其实用性仍有一定的局限.并且极大多数导向基导向的C—H键卤化反应发生在邻位的C—H键.因此, 设计新型的催化剂体系, 发展多样的选择性C—H键卤化反应类型仍是该领域长期的目标.通过发展底物本身官能团导向, 丰富反应底物类型, 使之能应用于天然产物、功能材料以及药物分子的合成中, 这无疑都具有极其重要的学术和应用价值.
-
-
[1]
Butler, A.; Walker, J. V. Chem. Rev. 1993, 93, 1937. doi: 10.1021/cr00021a014
-
[2]
Wan, X.-B.; Ma, Z.-X.; Li, B.-J.; Zhang, K.-Y.; Cao, S.-K.; Zhang, S.-W.; Shi, Z.-J. J. Am. Chem. Soc. 2006, 128, 7416. doi: 10.1021/ja060232j
-
[3]
Sotomayor, N.; Lete, E. Curr. Org. Chem. 2003, 7, 275. doi: 10.2174/1385272033372987
-
[4]
Dekker, M. Handbook of Grignard Reagents, Eds.: Silverrman, G. S.; Rakita, P. E., Marcel Dekker, Inc., New York, 1996, p. 736.
-
[5]
Levin, R. H. React. Intermed. 1985, 3, 1.
-
[6]
Crampton, M. R. Organic Reaction Mechanisms, Wiley, New York, 2003, p. 275.
-
[7]
(a) Yu, S. -C. ; Ma, S. -M. Chin. J. Org. Chem. 2002, 22, 307 (in Chinese).
(余世超, 麻生明, 有机化学, 2002, 22, 307. )
(b) Li, B. -J. ; Yang, S. -D. ; Shi, Z. -J. Synlett 2008, 7, 949.
(c) Li, C. -J. Acc. Chem. Res. 2009, 42, 335.
(d) Ackermann, L. Chem. Rev. 2011, 111, 1315.
(e) Wang, Y. ; Cheng, G. -L. ; Cui, X. Chin. J. Org. Chem. 2012, 32, 2018 (in Chinese).
(王勇, 程国林, 崔秀, 有机化学, 2012, 32, 2018. )
(f) Zhang, B. ; Guan, H. -X. ; Liu, B. ; Shi, B. -F. Chin. J. Org. Chem. 2014, 34, 1487 (in Chinese).
(张博, 管晗曦, 刘斌, 史炳锋, 有机化学, 2014, 34, 1487. )
(g) Gang, F. -L. ; Xu, G. -L. ; Dong, T. -S. ; Yang, L. ; Du, Z. -Y. Chin. J. Org. Chem. 2015, 35, 1428 (in Chinese).
(刚芳莉, 徐光利, 董涛生, 杨丽, 杜正银, 有机化学, 2015, 35, 1428. )
(h) Sun, C. -L. ; Shi, Z. -J. Chem. Rev. 2014, 114, 9219. -
[8]
(a) Kuwabe, S. ; Torraca, K. E. ; Buchwald, S. L. J. Am. Chem. Soc. 2001, 123, 12202.
(b) Beletskaya, I. P. ; Cheprakov, A. V. Coord. Chem. Rev. 2004, 248, 2337.
(c) Deng, W. ; Liu. L. ; Guo, Q. -X. Chin. J. Org. Chem. 2004, 24, 150 (in Chinese).
(邓维, 刘磊, 郭庆祥, 有机化学, 2004, 24, 150. )
(d) Corbet, J. P. ; Mignani, G. Chem. Rev. 2006, 106, 2651.
(e) Xu, H. -J. ; Tong, Q. -S. ; Lin, Y. -C. ; Li, Y. -Y. ; Feng, Y. -S. Chin. J. Org. Chem. 2010, 30, 9 (in Chinese).
(许华建, 蔄秋石, 林义成, 李源源, 冯乙巳, 有机化学, 2010, 30, 9. )
(f) Cheng, Y. -J. ; Sun, L. -P. Chin. J. Org. Chem. 2013, 33, 877 (in Chinese).
(成宜娟, 孙丽萍, 有机化学, 2013, 33, 877. )
(g) Dong, Q. -Z. ; Xiang, J. -N. ; Guo, C. -C. Chin. J. Org. Chem. 2014, 34, 147 (in Chinese).
(董奇志, 向建南, 郭灿城, 有机化学, 2014, 34, 147. )
(h) Ruiz-Castillo, P. ; Buchwald, S. L. Chem. Rev. 2016, 116, 12564.
(i) Wei, W. -T. ; Zhu, W. -M. ; Wu, Y. ; Huang, Y. -L. ; Liang, H. -Z. Chin. J. Org. Chem. 2017, 37, 1916 (in Chinese).
(魏文廷, 朱文明, 吴益, 黄依铃, 梁洪泽, 有机化学, 2017, 37, 1916. ) -
[9]
(a) De la Mare, P. B. Electrophilic Halogenation, Cambridge University Press, Cambridge, 1976, Chapter 5.
(b) Merkushev, E. B. Synthesis 1988, 923. -
[10]
(a) Dewkar, G. K.; Narina, S. V.; Sudalai, A. Org. Lett. 2003, 5, 4501.
(b) Tanemura, K.; Suzuki, T.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. Chem. Lett. 2003, 32, 932.
(c) Zhang, Y.; Shibatomi, K.; Yamamoto, H. Synlett 2005, 2837.
(d) Prakash, G. K. S.; Mathew, T.; Hoole, D.; Esteves, P. M.; Wang, Q.; Rasul, G.; Olah, G. A. J. Am. Chem. Soc. 2004, 126, 15770. -
[11]
Snieckus, V. Chem. Rev. 1990, 90, 879. doi: 10.1021/cr00104a001
-
[12]
Podgorsek, A.; Zupan, M.; Iskra, J. Angew. Chem., Int. Ed. 2009, 48, 8424. doi: 10.1002/anie.v48:45
-
[13]
Hao, W.; Liu, Y. Beilstein J. Org. Chem. 2015, 11, 2132. doi: 10.3762/bjoc.11.230
-
[14]
廖港, 史炳锋, 化学学报, 2015, 73, 1283. doi: 10.3866/PKU.WHXB201504291Liao, G.; Shi, B.-F. Acta Chim. Sinica 2015, 73, 1283(in Chinese). doi: 10.3866/PKU.WHXB201504291
-
[15]
Sloan, N. L.; Sutherland, A. Synthesis 2016, 48, 2969. doi: 10.1055/s-0035-1562439
-
[16]
Koval, I. V. Russ. J. Org. Chem. 2002, 38, 301. doi: 10.1023/A:1016390721218
-
[17]
(a) Wierlacher, S.; Sander, W.; Liu, M. T. H. J. Am. Chem. Soc. 1993, 115, 8943.
(b) Meddour, A.; Courtieu, J. Tetrahedron: Asymmetry 2000, 11, 3635. -
[18]
Dick, A. R.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2004, 126, 2300. doi: 10.1021/ja031543m
-
[19]
(a) Kalyani, D.; Dick, A. R.; Anani, W. Q.; Sanford, M. S. Org. Lett. 2006, 8, 2523.
(b) Kalyani, D.; Dick, A. R.; Anani, W. Q.; Sanford, M. S. Tetrahedron 2006, 62, 11483. -
[20]
Stowers, K. J.; Sanford, M. S. Org. Lett. 2009, 11, 4584. doi: 10.1021/ol901820w
-
[21]
Bedford, R. B.; Haddow, M. F.; Mitchell, C. J.; Webster, R. L. Angew. Chem., Int. Ed. 2011, 50, 5524. doi: 10.1002/anie.201101606
-
[22]
(a) Giri, R.; Lam, J. K.; Yu, J. Q. J. Am. Chem. Soc. 2010, 132, 686.
(b) Tobisu, M.; Ano, Y.; Chatani, N. Org. Lett. 2009, 11, 3250. -
[23]
Sadhu, P.; Alla, S. K.; Punniyamurthy, T. J. Org. Chem. 2013, 78, 6104. doi: 10.1021/jo400755q
-
[24]
Sun, M.; Chen, X.-X.; Zhang, L.; Sun, W.; Wang, Z.; Guo, P.-Y.; Li, Y.-M.; Yang, X.-J. Org. Biomol. Chem. 2016, 14, 323. doi: 10.1039/C5OB02153G
-
[25]
Zhao, J.-L.; Chen, X.-X.; Xie, H.; Ren, J.-T.; Gou, X.-F.; Sun, M. Synlett 2017, 28, 1232. doi: 10.1055/s-0036-1588756
-
[26]
Pron, F.; Fossey, C.; Santos, J. S. O.; Cailly, T.; Fabis, F. Chem. Eur. J. 2014, 20, 1. doi: 10.1002/chem.201390210
-
[27]
Du, B.; Jiang, X.-Q.; Sun, P.-P. J. Org. Chem. 2013, 78, 2786. doi: 10.1021/jo302765g
-
[28]
Sarkar, D.; Melkonyan, F. S.; Gulevich, A. V.; Gevorgyan, V. Angew. Chem., Int. Ed. 2013, 52, 1. doi: 10.1002/anie.201209858
-
[29]
Purohit, V. B.; Karad, S. C.; Patel, K. H.; Raval, D. K. Catal. Sci. Technol. 2015, 5, 3113. doi: 10.1039/C5CY00137D
-
[30]
Gao, D.-W.; Gu, Q.; You, S.-L. ACS Catal. 2014, 4, 2741. doi: 10.1021/cs500813z
-
[31]
(a) Denmark, S. E.; Fan, Y. J. Am. Chem. Soc. 2002, 124, 4233.
(b) Malkov, A. V.; Dufková, L.; Farrugia, L.; Kočovský, P. Angew. Chem., Int. Ed. 2003, 42, 3674. -
[32]
Gutnov, A.; Heller, B.; Fischer, C.; Drexler, H. J.; Spannenberg, A.; Sundermann, B.; Sundermann, C. Angew. Chem., Int. Ed. 2004, 43, 3795. doi: 10.1002/(ISSN)1521-3773
-
[33]
Schröder, N.; Wencel-Delord, J.; Glorius, F. J. Am. Chem. Soc. 2012, 134, 8298. doi: 10.1021/ja302631j
-
[34]
Schröder, N.; Lied, F.; Glorius, F. J. Am. Chem. Soc. 2015, 137, 1448. doi: 10.1021/jacs.5b00283
-
[35]
Hwang, H.; Kim, J.; Jeong, J.; Chang, S. J. Am. Chem. Soc. 2014, 136, 10770. doi: 10.1021/ja5053768
-
[36]
Ding, Q.; Zhou, X.; Pu, S.; Cao, P. Tetrahedron 2015, 71, 2376. doi: 10.1016/j.tet.2015.03.004
-
[37]
(a) Zhang, C. ; Tang, C. ; Jiao, N. Chem. Soc. Rev. 2012, 41, 3464.
(b) King, A. E. ; Huffman, L. M. ; Casitas, A. ; Costas, M. ; Ribas, X. ; Stahl, S. S. J. Am. Chem. Soc. 2010, 132, 12068.
(c) Wendlandt, A. E. ; Suess, A. M. ; Stahl, S. S. Angew. Chem. , Int. Ed. 2011, 50, 11062.
(d) Liu, W. ; Bi, Y. -L. Chin. J. Org. Chem. 2012, 32, 1041 (in Chinese).
(刘伟, 毕艳兰, 有机化学, 2012, 32, 1041. )
(e) Liu, C. ; Liu, D. ; Lei, A. Acc. Chem. Res. 2014, 47, 3459.
(f) Liu, Y. -Y. ; Zhang, W. -B. Chin. J. Org. Chem. 2016, 36, 2249 (in Chinese).
(刘媛媛, 张万斌, 有机化学, 2016, 36, 2249. ) -
[38]
Du, Z.-J.; Gao, L.-X.; Lin, Y.-J.; Han, F.-S. ChemCatChem 2014, 6, 123. doi: 10.1002/cctc.v6.1
-
[39]
Zhang, C.; Tang, C.; Jiao, N. ChemSocRev 2012, 41, 3464. http://www.ncbi.nlm.nih.gov/pubmed/22349590
-
[40]
Urones, B.; Martínez, A. M.; Rodríguez, N.; Arrayás, R. G.; Carretero, J. C. Chem. Commun. 2013, 49, 11044. doi: 10.1039/c3cc47174h
-
[41]
Li, B.; Liu, B.; Shi, B.-F. Chem. Commun. 2015, 51, 5093. doi: 10.1039/C5CC00531K
-
[42]
Chinnagolla, R. K.; Pimparkar, S.; Jeganmohan, M. Chem. Commun. 2013, 49, 3146. doi: 10.1039/c3cc41124a
-
[43]
Wang, L.; Ackermann, L. Chem. Commun. 2014, 50, 1083. doi: 10.1039/C3CC47852A
-
[44]
Warratz, S.; Burns, D. J.; Zhu, C.-J.; Korvorapun, K.; Rogge, T.; Scholz, J.; Jooss, C.; Gelman, D.; Ackermann, L. Angew. Chem., Int. Ed. 2017, 56, 1557. doi: 10.1002/anie.201609014
-
[45]
Yu, D.-G.; Gensch, T.; de Azambuja, F.; Vásquez-Céspedes, S.; Glorius, F. J. Am. Chem. Soc. 2014, 136, 17722. doi: 10.1021/ja511011m
-
[46]
Wang, T.-H.; Guan, X.-L.; Lu, H.-Y.; Liu, Z.-W.; Ji, M. Appl. Surf. Sci. 2017, 398, 1. doi: 10.1016/j.apsusc.2016.11.180
-
[47]
Pal, P.; Singh, H.; Panda, A. B.; Ghosh, S. C. Asian J. Org. Chem. 2015, 4, 879. doi: 10.1002/ajoc.201500245
-
[1]
-

图式2 环钯中间体形成可能机理
Scheme 2 Proposed mechanism for the formation of cyclopalladated complexes

图式3 天然化合物Paucifloral F和iso-Paucifloral F前体的合成
Scheme 3 Synthesis of paucifloral F and iso-paucifloral F precursors
-


 下载:
下载:








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 5292
- HTML全文浏览量: 355
 下载:
下载:
