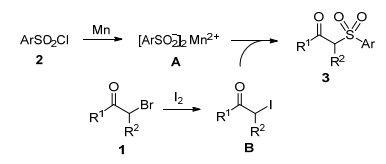
Scheme 1.
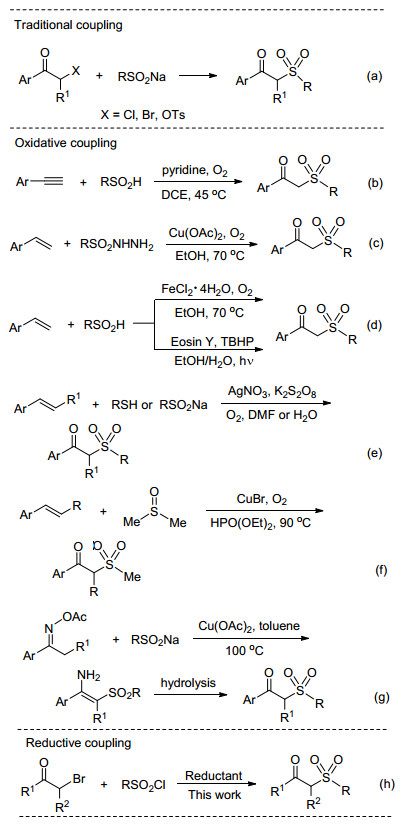
Different synthetic approaches to β-ketosulfones
Facile Access to β-Ketosulfones via Mn-Mediated Reductive Coupling of α-Bromoketones with Sulfonyl Chlorides
Zhen Chen , Kang Guo , Rongshun Chen , Chen Gu , Huating Zhou , Yingguang Zhu

The sulfonyl group as a key structural motif is ubiquitous in many bioactive compounds and marketed drugs.[1] Among the sulfonyl group-containing molecules, β-ketosul-fones are valuable and highly desirable synthetic targets due to their unique biological properties such as antifungal and antibacterial activities.[2] In addition, they are also important building blocks that can be converted into various com-pounds such as β-hydroxysulfones, [3] vinylsulfones, [4] α-halo methylsulfones, [5] acetylenes, [6] allenes, [7] quino-lines, [8] and polyfunctionalized 4H-pyrans, [9] etc. Thus, great efforts have been made for the synthesis of β-ketosulfones.
The conventional synthetic approaches to β-ketosulfones are mainly focused on the traditional coupling of sulfinates with phenacyl halides or tosylates (Scheme 1a).[10] However, some of these methods suffer from certain disa-dvantages such as relatively harsh reaction conditions and narrow substrate scope.
Recently, an efficient synthesis of β-ketosulfones has been achieved via the novel oxidative coupling of arylalkynes or arylalkenes with sulfonylating agents. For example, in 2013, Lei and co-workers[11a] reported an elegant method for the preparation of β-ketosulfones from alkynes and sulfinic acids, in which dioxygen was used as the green oxidant and oxygen source (Scheme 1b). In the same year, Wang's group[12] developed a highly efficient copper-catalyzed oxidative coupling reaction of alkenes and sulfonylhydrazides toward β-ketosulfones (Scheme 1c). They[13, 14] subsequently also demonstrated that β-keto-sulfones could be obtained from olefins and sulfinic acids through iron-catalyzed or visible-light-induced oxidative sulfonylation (Scheme 1d). With thiophenols as the sulfonylation precursors and alkenes as the substrates in the presence of AgNO3/K2S2O8, an efficient synthetic approach to β-ketosulfones was achieved by Yadav and co-workers (Scheme 1e).[15] The authors[16] also reported the synthesis of β-ketosulfones from arenesulfinate salts and olefins with AgNO3/K2S2O8 catalytic system (Scheme 1e). Loh's group[17] disclosed a new oxysulfonylation of alkenes using dimethyl sulfoxide (DMSO) as the methyl sulfonyl source and CuBr as the catalyst under O2 atmosphere (Scheme 1f). In 2014, Jiang et al.[18] developed a novel copper-catalyzed oxidative coupling of oxime acetates and sodium sulfinates to afford β-ketosulfones (Scheme 1g). In this process, no external oxidants were required. Despite recent great progress, practical, mild, and efficient methods for the synthesis of β-ketosulfones utilizing commercially available sulfonyla-ting agents remain highly desirable and valuable. Herein, we report a Mn-mediated[19] reductive coupling of α-bromo-ketones with sulfonyl chlorides[20] to access various β-alkyl and β-aryl-β-ketosulfones (Scheme 1h). The present method features mild conditions, high yields, broad substrate scope, direct utilization of cheap and commercially available sulfonyl source, and gram-scale synthesis.
Our investigation commenced with α-bromoketone 1a and p-toluenesulfonyl chloride (2a) as the model substrates. Gratifyingly, the desired coupling product 3a could be isolated in 20% yield when 1a and 2a were treated with iron powder in N, N-dimethylacetamide (DMA) at room temperature for 24 h (Table 1, Entry 1). Encouraged by this initial result, we next focused our attention on the optimization of reaction conditions. Among the reductants examined, manganese was found to be the best and afforded prodcut 3a in 52% yield (Table 1,Entry 3). The effect of additives on the yield was subsequently investigated. The addition of additives (10 mol%) including NaI, tetrabuty-lammonium iodide (TBAI), I2, MgCl2, trimethylsilyl chloride (TMSCl), and trifluoroacetic acid (TFA) increased the yield of 3a more or less (Table 1, Entries 4~9). When 10 mol% I2 was used as an additive, the highest yield (86%) could be obtained (Table 1, Entry 6). Other solvents such as DMF, 1, 3-dimethylpropyleneurea (DMPU), N-methyl-2-pyrrolidone (NMP), and DMSO resulted in an inferior yield than DMA (Table 1, Entry 6 and Entries 10~13). No product was detected when THF or CH3CN was used (Table 1, Entries 14 and 15).
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | Reductant | Additive | Solvent | Yieldb/% |
| 1 | Fe | none | DMA | 20 |
| 2 | Zn | none | DMA | trace |
| 3 | Mn | none | DMA | 52 |
| 4 | Mn | NaI | DMA | 58 |
| 5 | Mn | TBAI | DMA | 65 |
| 6 | Mn | I2 | DMA | 86 |
| 7 | Mn | MgCl2 | DMA | 56 |
| 8 | Mn | TMSCl | DMA | 68 |
| 9 | Mn | TFA | DMA | 61 |
| 10 | Mn | I2 | DMF | 59 |
| 11 | Mn | I2 | DMPU | 73 |
| 12 | Mn | I2 | NMP | 57 |
| 13 | Mn | I2 | DMSO | 15 |
| 14 | Mn | I2 | THF | 0 |
| 15 | Mn | I2 | CH3CN | 0 |
| a All reactions were carried out with 1a (0.20 mmol), 2a (0.30 mmol), reductant (0.60 mmol), and additive (0.020 mmol) in solvent (0.80 mL) at room temperature for 24 h. DMA=N, N-dimethylacetamide, TBAI=tetrabutylammonium iodide, TMSCl=trimethylsilyl chloride, TFA=tri-fluoroacetic acid, DMPU=1, 3-dimethylpropyleneurea, NMP=N-methyl-2-pyr-rolidone. b Isolated yield based on 1a. | ||||
With the optimal reaction conditions in hand, the generality of this reductive coupling was subsequently investigated with various α-bromoketones. As shown in Table 2, a wide range of α-bromoketones can be reductively coupled with p-toluenesulfonyl chloride (2a) to give the corresponding β-ketosulfones in 63%~92% yield. For aromatic α-bromoketones bearing both electron-donating and electron-withdrawing groups on the phenyl ring, the reaction proceeded smoothly and afforded the coupling products (3a~3g) in good to excellent yields. Naphthyl and thienyl-substituted α-bromoketones were also effective substrates, giving the corresponding β-ketosulfones 3h and 3i in 78% and 73% yields, respectively. Remarkably, this present coupling reaction can be extended to acyclic and cyclic aliphatic α-bromoketones to provide β-alkyl-β-ketosulfones (3j~3m) in 63%~81% yield.
下载:
导出CSV
 |
||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
||
| a All reactions were carried out with 1 (0.20 mmol), 2a (0.30 mmol), Mn (0.60 mmol), and I2 (0.020 mmol) in DMA (0.80 mL) at room temperature for 24 h. Isolated yield based on 1. | ||
The scope of sulfonyl chlorides was also investigated with 1a under the optimized reaction conditions, and the results are summarized in Table 3. Various arylsulfonyl chlorides bearing substituents (such as Cl, NHAc, or OMe) on the para-position of aromatic ring afforded the products 3n~3q in good yields. 3-Methylbenzenesulfonyl chloride was suitable coupling partner to give the product 3r in 75% yield. Naphthyl and thienyl were also tolerated, lead-ing to the corresponding products (3s and 3t) in 70% and 93% yields, respectively. Unfortunately, propylsulfonyl chloride, an alkyl-substituted sulfonyl chloride, failed to provide the desired product 3u under the standard conditions.
下载:
导出CSV
 |
|
 |
 |
 |
 |
 |
 |
 |
 |
| a All reactions were carried out with 1a (0.20 mmol), 2 (0.30 mmol), Mn (0.60 mmol), and I2 (0.020 mmol) in DMA (0.80 mL) at room temperature for 24 h. Isolated yield based on 1a. | |
To demonstrate the synthetic utility of this method, gram-scale synthesis of β-ketosulfone 3e was conducted in an open flask under standard reaction conditions (Eq. 1). This transformation proceeded smoothly to furnish the desired coupling product 3e (4.11 g, 89% yield). Notably, no substantial drop in the yield was observed during the gram-scale synthesis.
|
|
(1) |
A plausible mechanism for this reductive coupling of α-bromoketones with sulfonyl chlorides is proposed in Scheme 2.[20] Sulfonyl chloride 2 is first reduced by manganese to generate the corresponding sulfonyl anion A. More reactive α-iodoketone B is likely formed in situ from α-bromoketone 1 and I2 via a halogen exchange reaction.[21, 22] Intermediate A then nucleophilically attacks the in situ generated α-iodoketone B to deliver the desired products 3.
In summary, we have developed a mild and efficient synthetic approach to β-ketosulfones via Mn-mediated reductive coupling of α-bromoketones with cheap and commercially available sulfonyl chlorides. Various aliphatic and aromatic α-bromoketones can be effectively coupled with sulfonyl chlorides to give the corresponding β-alkyl and β-aryl-β-ketosulfones in high yields (up to 93%). This coupling reaction exhibits a broad substrate scope and good functional group tolerance. The present protocol is mild, operationally simple, and amenable to the gram scale.Future efforts will focus on the development of other coupling processes and the investigation of their applications in organic synthesis.
All commercially available reagents were used without further purification. Column chromatography was performed on silica gel (200~300 mesh). 1H NMR and 13C NMR spectra were recorded on a 400 MHz spectrometer operating at 400 and 100 MHz, respectively. High resolution mass spectra (HRMS) were recorded on a Bruker MicroTOF ESI mass spectrometer. IR spectra were recorded on a FT-IR spectrometer. Melting points were uncorrected. Aromatic α-bromoketones were prepared via bromination of the commercially available ketones according to the literature procedure.[23] Aliphatic α-bromoke-tones were synthesized via bromination of the commercially available ketones following the reported methods.[24]
To a reaction tube equipped with a magnetic stir bar were added α-bromoketone (0.20 mmol), sulfonyl chloride (0.30 mmol), Mn (32.9 mg, 0.60 mmol, 325 mesh), I2 (5.1 mg, 10 mol%, 0.020 mmol), and DMA (0.80 mL). The resulting mixture was vigorously stirred at room temperature for 24 h. After the reaction was completed, water (10 mL) was added and the mixture was extracted with ethyl acetate (10 mL×3). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel using petroleum ether/ethyl acetate as the eluent to give the pure product 3.
1-Phenyl-2-tosylpropan-1-one (3a):[25] White solid (49.7 mg, 86%), m.p. 99~100 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J=7.8 Hz, 2H), 7.65 (d, J=8.1 Hz, 2H), 7.59 (t, J=7.4 Hz, 1H), 7.46 (t, J=7.8 Hz, 2H), 7.29 (d, J=8.1 Hz, 2H), 5.18 (q, J=6.9 Hz, 1H), 2.41 (s, 3H), 1.55 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.5, 145.2, 136.1, 133.8, 132.9, 129.6, 129.4, 129.0, 128. 6, 64.7, 21.5, 13.0; IR (KBr) v: 1676, 1595, 1450, 1314, 1239, 1140 cm-1.
1-Phenyl-2-tosylethanone (3b):[25, 26] White solid (43.7 mg, 80%), m.p. 104~105 ℃ (lit. m.p. 105~106 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.94 (d, J=7.7 Hz, 2H), 7.76 (d, J=8.1 Hz, 2H), 7.61 (t, J=7.4 Hz, 1H), 7.47 (t, J=7.7 Hz, 2H), 7.33 (d, J=8.1 Hz, 2H), 4.73 (s, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 188.1, 145.3, 135.6, 134.2, 129.8, 129.2, 128.7, 128.5, 63.4, 21.6; IR (KBr) v: 1679, 1596, 1447, 1319, 1271, 1151 cm-1.
1-(p-Tolyl)-2-tosylpropan-1-one (3c): White solid (43.5 mg, 72%), m.p. 106~107 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.88 (d, J=7.9 Hz, 2H), 7.65 (d, J=7.9 Hz, 2H), 7.29 (dd, J=11.8, 8.1 Hz, 4H), 5.13 (q, J=6.9 Hz, 1H), 2.43 (s, 6H), 1.54 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.0, 145.2, 145.1, 133.8, 133.0, 129.8, 129.43, 129.38, 129.3, 64.8, 21.7, 21.6, 13.2; IR (KBr) v: 1672, 1608, 1375, 1312, 1246, 1200, 1142 cm-1; HRMS (ESI-TOF) calcd for C17H19O3S [M+H]+ 303.1049, found 303.1044.
1-(p-Tolyl)-2-tosylethanone (3d):[25, 26] White solid (39.6 mg, 69%), m.p. 113~114 ℃ (lit. m.p. 114~116 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.85 (d, J=8.0 Hz, 2H), 7.76 (d, J=8.0 Hz, 2H), 7.33 (d, J=8.0 Hz, 2H), 7.27 (d, J=8.0 Hz, 2H), 4.69 (s, 2H), 2.44 (s, 3H), 2.42 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 187.6, 145.5, 145.2, 135.7, 133.3, 129.8, 129.5, 129. 5, 128.5, 63.5, 21.73, 21.66; IR (KBr) v: 2950, 2908, 1687, 1605, 1406, 1316, 1205, 1141 cm-1.
1-(4-Chlorophenyl)-2-tosylethanone (3e):[25] White solid (56.9 mg, 92%, 0.2 mmol scale; 4.13 g, 89%, 15 mmol scale), m.p. 136~137 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.90 (d, J=8.6 Hz, 2H), 7.74 (d, J=8.2 Hz, 2H), 7.45 (d, J=8.6 Hz, 2H), 7.34 (d, J=8.2 Hz, 2H), 4.69 (s, 2H), 2.45 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 187.0, 145.5, 141.0, 135.4, 134.0, 130.7, 129.9, 129.1, 128.5, 63.6, 21.7; IR (KBr) v: 1679, 1590, 1315, 1278, 1148 cm-1.
1-(3-Chlorophenyl)-2-tosylethanone (3f):[25] White solid (49.8 mg, 81%), m.p. 124~126 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.87~7.83 (m, 2H), 7.75 (d, J=8.2 Hz, 2H), 7.60~7.56 (m, 1H), 7.44 (t, J=8.2 Hz, 1H), 7.35 (d, J=8.0 Hz, 2H), 4.70 (s, 2H), 2.45 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 187.0, 145.6, 137.1, 135.4, 135.1, 134.2, 130.1, 129.9, 129.1, 128.5, 127.5, 63.6, 21.7; IR (KBr) v: 1681, 1653, 1593, 1429, 1315, 1300, 1259, 1149, 1085 cm-1.
1-(2-Chlorophenyl)-2-tosylethanone (3g): White solid (40.6 mg, 66%), m.p. 101~103 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.76 (d, J=8.2 Hz, 2H), 7.57~7.53 (m, 1H), 7.46~7.41 (m, 1H), 7.40~7.30 (m, 4H), 4.82 (s, 2H), 2.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 190.3, 145.3, 137.3, 135.9, 133.1, 131.5, 130.6, 130.6, 129.8, 128.5, 127.2, 66.4, 21.7; IR (KBr) v: 1697, 1586, 1435, 1310, 1141, 1084 cm-1; HRMS (ESI-TOF) calcd for C15H14ClO3S [M+H]+ 309.0347, found 309.0349.
1-(Naphthalen-2-yl)-2-tosylethanone (3h):[27] White solid (50.4 mg, 78%), m.p. 148~149 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.44 (s, 1H), 7.95 (d, J=8.2 Hz, 2H), 7.90~7.84 (m, 2H), 7.77 (d, J=8.2 Hz, 2H), 7.63 (t, J=7.4 Hz, 1H), 7.57 (t, J=7.4 Hz, 1H), 7.30 (d, J=8.2 Hz, 2H), 4.85 (s, 2H), 2.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 188.0, 145.4, 135.9, 135.6, 133.0, 132.2, 132.1, 129.9, 129.8, 129.3, 128.7, 128.6, 127.7, 127.1, 123.9, 63.7, 21.6; IR (KBr) v: 1667, 1595, 1309, 1290, 1148, 1083 cm-1.
1-(Thiophen-2-yl)-2-tosylethanone (3i):[25] White solid (40.9 mg, 73%), m.p. 109~110 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.81 (d, J=3.8 Hz, 1H), 7.79~7.73 (m, 3H), 7.34 (d, J=8.1 Hz, 2H), 7.16 (t, J=4.4 Hz, 1H), 4.62 (s, 2H), 2.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 180.3, 145.4, 143.1, 136.4, 135.4, 135.3, 129.8, 128.7, 128.5, 64.6, 21.7; IR (KBr) v: 2925, 1653, 1415, 1328, 1314, 1283 cm-1.
2-Tosylpentan-3-one (3j): White solid (36.1 mg, 75%), m.p. 70~71 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.66 (d, J=8.2 Hz, 2H), 7.36 (d, J=8.2 Hz, 2H), 4.16 (q, J=7.0 Hz, 1H), 3.03~2.91 (m, 1H), 2.68~2.58 (m, 1H), 2.46 (s, 3H), 1.39 (d, J=7.0 Hz, 3H), 1.07 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 203.0, 145.4, 133.1, 129.7, 129.3, 69.8, 37.2, 21.6, 12.0, 7.4; IR (KBr) v: 1720, 1595, 1456, 1405, 1315, 1150, 1085 cm-1; HRMS (ESI-TOF) calcd for C12H16NaO3S [M+Na]+ 263.0712, found 263.0726.
3, 3-Dimethyl-1-tosylbutan-2-one (3k): White solid (40.1 mg, 77%), m.p. 112~113 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.82 (d, J=8.3 Hz, 2H), 7.36 (d, J=8.3 Hz, 2H), 4.31 (s, 2H), 2.45 (s, 3H), 1.12 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 203.4, 145.0, 136.4, 129.7, 128.6, 60.7, 45.2, 25.5, 21.7; IR (KBr) v: 2952, 2908, 1717, 1596, 1479, 1369, 1320, 1390, 1181, 1139, 1056 cm-1; HRMS (ESI-TOF) calcd for C13H19O3S [M+H]+ 255.1049, found 255.1045.
2-Tosylcyclopentanone (3l):[28] Colorless oil (30.1 mg, 63%). 1H NMR (400 MHz, CDCl3) δ: 7.76 (d, J=8.2 Hz, 2H), 7.37 (d, J=8.2Hz, 2H), 3.77~3.70 (m, 1H), 2.67~2.61 (m, 1H), 2.46 (s, 3H), 2.43~2.35 (m, 2H), 2.34~2.14 (m, 2H), 1.94~1.84 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 207.3, 145.2, 135.0, 129.7, 129.0, 69.4, 38.7, 25.0, 21.7, 20.0; IR (film) v: 1748, 1597, 1403, 1303, 1149, 1084 cm-1.
2-Tosylcyclohexanone (3m):[29, 30] White solid (40.8 mg, 81%), m.p. 80~82 ℃ (lit. m.p. 76~80 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.76 (d, J=8.3 Hz, 2H), 7.35 (d, J=8.3Hz, 2H), 3.82 (t, J=5.0 Hz, 1H), 2.85~2.76 (m, 1H), 2.57~2.50 (m, 1H), 2.46~2.39 (m, 1H), 2.45(s, 3H), 2.22 (m, 2H), 2.01 (m, 1H), 1.86~1.72 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 202.4, 145.1, 135.1, 129.7, 128.8, 72.8, 41.5, 27.5, 26.4, 21.8, 21.6; IR (KBr) v: 1716, 1598, 1443, 1346, 1321, 1267, 1144, 1093, 1059 cm-1.
1-Phenyl-2-(phenylsulfonyl)propan-1-one (3n):[11a] White solid (39.6 mg, 72%), m.p. 83~84 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.96 (d, J=7.4 Hz, 2H), 7.79 (d, J=7.3 Hz, 2H), 7.66~7.58 (m, 2H), 7.55~7.44 (m, 4H), 5.18 (q, J=6.9 Hz, 1H), 1.57 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.4, 136.0, 135.9, 134.2, 134.0, 129.7, 129.1, 128.8, 128.7, 64. 8, 13.1; IR (KBr) v: 1678, 1447, 1309, 1242, 1145 cm-1.
2-((4-Chlorophenyl)sulfonyl)-1-phenylpropan-1-one (3o): White solid (45.1 mg, 73%), m.p. 118~119 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J=7.6 Hz, 2H), 7.72 (d, J=8.5 Hz, 2H), 7.64 (t, J=7.4 Hz, 1H), 7.56~7.45 (m, 4H), 5.18 (q, J=6.9 Hz, 1H), 1.57 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.4, 141.1, 135.9, 134.3, 134.2, 131.3, 129.2, 129.1, 128.8, 65.0, 13.3; IR (KBr) v: 1676, 1596, 1449, 1320, 1155, 1082 cm-1; HRMS (ESI-TOF) calcd for C15H13ClNaO3S [M+Na]+ 331.0166, found 331.0165.
N-(4-((1-Oxo-1-phenylpropan-2-yl)sulfonyl)phenyl)ace-tamide (3p): White solid (52.2 mg, 79%), m.p. 184~185 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.01 (s, 1H), 7.63~7.58 (m, 2H), 7.37 (d, J=8.9 Hz, 2H), 7.29~7.21 (m, 3H), 7.08 (t, J=7.8 Hz, 2H), 5.27 (q, J=6.7 Hz, 1H), 1.69 (s, 3H), 0.99 (d, J=6.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 192.8, 169.3, 144.5, 136.2, 134.0, 130.5, 130.0, 129.3, 128.7, 118.4, 63.8, 24.3, 12.7; IR (KBr) v: 3365, 1696, 1677, 1593, 1526, 1403, 1322, 1294, 1211, 1132 cm-1; HRMS (ESI-TOF) calcd for C17H18NO4S [M+H]+ 332.0951, found 332.0948.
2-((4-Methoxyphenyl)sulfonyl)-1-phenylpropan-1-one (3q): White solid (54.1 mg, 89%), m.p. 99~100 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.01~7.95 (m, 2H), 7.73~7.67 (m, 2H), 7.61 (t, J=7.4 Hz, 1H), 7.48 (t, J=7.7 Hz, 2H), 6.97 (t, J=5.9 Hz, 2H), 5.16 (q, J=6.9 Hz, 1H), 3.86 (s, 3H), 1.55 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.7, 164.1, 136.2, 134.0, 131.9, 129.1, 128.7, 127.2, 114.0, 64.9, 55.6, 13.2; IR (KBr) v: 1668, 1591, 1447, 1344, 1202, 1136 cm-1; HRMS (ESI-TOF) calcd for C16H16NaO4S [M+Na]+ 327.0662, found 327.0659.
1-Phenyl-2-(m-tolylsulfonyl)propan-1-one (3r): Color-less oil (43.0 mg, 75%). 1H NMR (400 MHz, CDCl3) δ: 7.95 (d, J=7.5 Hz, 2H), 7.62~7.56 (m, 3H), 7.49~7.37 (m, 4H), 5.17 (q, J=6.9 Hz, 1H), 2.39 (s, 3H), 1.58 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.4, 139.1, 136.1, 135.9, 135.0, 134.0, 129.9, 129.0, 128.72, 128.65, 126.8, 64.8, 21.2, 13.0; IR (film) v: 1682, 1596, 1449, 1303, 1236, 1143, 1082 cm-1; HRMS (ESI-TOF) calcd for C16H16NaO3S [M+Na]+ 311.0712, found 311.0718.
2-(Naphthalen-2-ylsulfonyl)-1-phenylpropan-1-one (3s): White solid (45.3 mg, 70%), m.p. 113~115 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.36 (s, 1H), 7.99~7.89 (m, 5H), 7.76 (dd, J=8.6, 1.6 Hz, 1H), 7.67 (t, J=7.4 Hz, 1H), 7.64~7.53 (m, 2H), 7.43 (t, J=7.7 Hz, 2H), 5.25 (q, J=6.9 Hz, 1H), 1.61 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.4, 136.1, 135.4, 134.0, 133.0, 131.9, 131.8, 129.48, 129.46, 129.1, 129.0, 128.7, 127.9, 127.6, 124.0, 65.0, 13.2; IR (KBr) v: 1675, 1593, 1446, 1313, 1143, 1128 cm-1; HRMS (ESI-TOF) calcd for C19H17O3S [M+H]+ 325.0893, found 325.0896.
1-Phenyl-2-(thiophen-2-ylsulfonyl)propan-1-one (3t): Viscous light yellow oil (51.9 mg, 93%). 1H NMR (400 MHz, CDCl3) δ: 8.02~7.95 (m, 2H), 7.75 (dd, J=5.0, 1.2 Hz, 1H), 7.65~7.57 (m, 2H), 7.49 (t, J=7.7 Hz, 2H), 7.13 (dd, J=5.0, 3.9 Hz, 1H), 5.25 (q, J=6.9 Hz, 1H), 1.66 (d, J=6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.3, 136.4, 136.3, 135.9, 135.3, 134.1, 129.0, 128.8, 127.8, 65.3, 13.3; IR (film) v: 1678, 1595, 1449, 1401, 1319, 1144, 731 cm-1; HRMS (ESI-TOF) calcd for C13H12NaO3S2 [M+ Na]+ 303.0120, found 303.0113.
Supporting Information Copies of 1H NMR and 13C NMR spectra of the products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
For selected exam. p. les, see: (a) Napier, C. ; Stewart, M. ; Melrose, H. ; Hopkins, B. ; McHarg, A. ; Wallis, R. Eur. J. Pharmacol. 1999, 375, 61.
(b) Petrov, K. G. ; Zhang, Y. ; Carter, M. ; Cockerill. G. S. ; Dickerson, S. ; Gauthier, C. A. ; Guo, Y. ; Mook, R. A. ; Rusnak, D. W. ; Walker, A. L. ; Wood, E. R. ; Lackey, K. E. Bioorg. Med. Chem. Lett. 2006, 16, 4686.
(c) Ma, M. ; Cheng, Y. ; Xu, Z. ; Xu, P. ; Qu, H. ; Fang, Y. ; Wen, L. Eur. J. Med. Chem. 2007, 42, 93.
(d) Jiang, J. ; Zou, H. ; Yi, N. ; Wang, R. ; Zhang, H. ; Lan, L. ; Xiang, J. Chin. J. Chem. 2016, 34, 1245.
(a) Wolf, W. M. J. Mol. Struct. 1999, 474, 113.
(b) Curti, C. ; Laget, M. ; Carle, A. O. ; Gellis, A. ; Vanelle, P. Eur. J. Med. Chem. 2007, 42, 880.
(c) Xiang, J. ; Ipek, M. ; Suri, V. ; Tam, M. ; Xing, Y. ; Huang, N. ; Zhang, Y. ; Tobin, J. ; Mansour, T. S. ; McKew, J. Bioorg. Med. Chem. 2007, 15, 4396.
(a) Bertus, P. ; Phansavath, P. ; Ratovelomanana-Vidal, V. ; Genêt, J. -P. ; Touati, A. R. ; Homri, T. ; Hassine, B. B. Tetrahedron: Asymmetry 1999, 10, 1369.
(b) Huang, X. ; Zhao, M. ; Li, N. ; Li, H. ; Li, J. ; Wang, X. Chin. J. Chem. 2014, 32, 803.
Sengupta, S.; Sarma, D. S.; Mondal, S. Tetrahedron:Asymmetry 1998, 9, 2311. doi: 10.1016/S0957-4166(98)00235-3
Suryakiran, N.; Reddy, T. S.; Suresh, V.; Lakshman, M.; Venkateswarlu, Y. Tetrahedron Lett. 2006, 47, 4319. doi: 10.1016/j.tetlet.2006.04.123
(a) Bartlett, P. A. ; Green, Ⅲ, F. R. ; Rose, E. H. J. Am. Chem. Soc. 1978, 100, 4852.
(b) Ihara, M. ; Suzuki, S. ; Taniguchi, T. ; Tokunaga, Y. ; Fukumoto, K. Tetrahedron 1995, 51, 9873.
Baldwin, J. E.; Adlington, R. M.; Crouch, N. P.; Hill, R. L.; Laffey, T. G. Tetrahedron Lett. 1995, 36, 7925. doi: 10.1016/0040-4039(95)01643-V
Swenson, R. E.; Sowin, T. J.; Zhang, H. Q. J. Org. Chem. 2002, 67, 9182. doi: 10.1021/jo0203387
Marco, J. L. J. Org. Chem. 1997, 62, 6575. doi: 10.1021/jo9705982
(a) Vennstra, G. E. ; Zwaneburg, B. Synthesis 1975, 519.
(b) Xie, Y. -Y., Chen, Z. -C. Synth. Commun. 2001, 31, 3145.
(a) Lu, Q. ; Zhang, J. ; Zhao, G. ; Qi, Y. ; Wang, H. ; Lei, A. J. Am. Chem. Soc. 2013, 135, 11481.
(b) Wang, H. ; Wang, G. ; Lu, Q. ; Chiang, C. -W. ; Peng, P. ; Zhou, J. ; Lei, A. Chem. Eur. J. 2016, 22, 14489.
Wei, W.; Liu, C.; Yang, D.; Wen, J.; You, J.; Suo, Y.; Wang, H. Chem. Commun. 2013, 49, 10239. doi: 10.1039/c3cc45803b
Wei, W.; Wen, J.; Yang, D.; Wu, M.; You, J.; Wang, H. Org. Biomol. Chem. 2014, 12, 7678. doi: 10.1039/C4OB01369G
Yang, D.; Huang B.; Wei, W.; Li, J.; Lin, G.; Liu, Y.; Ding, J.; Sun, P.; Wang, H. Green Chem. 2016, 18, 5630. doi: 10.1039/C6GC01403H
Singh, A. K.; Chawla, R.; Keshari, T.; Yadav, V. K.; Yadav, L. D. S. Org. Biomol. Chem. 2014, 12, 8550. doi: 10.1039/C4OB00776J
Singh, A. K.; Chawla, R.; Yadav, L. D. S. Tetrahedron Lett. 2014, 55, 4742. doi: 10.1016/j.tetlet.2014.06.086
Jiang, Y.; Loh, T.-P. Chem. Sci. 2014, 5, 4939. doi: 10.1039/C4SC01901F
Tang, X.; Huang, L.; Xu, Y.; Yang, J.; Wu, W.; Jiang, H. Angew. Chem., Int. Ed. 2014, 53, 4205. doi: 10.1002/anie.201311217
For selected reviews on transition metal-catalyzed carbon-carbon bond-forming reductive coupling with stoichiometric reductants such as Mn, see: (a) Knappke, C. E. I. ; Grupe, S. ; G rtner, D. ; Corpet, M. ; Gosmini, C. ; Jacobi von Wangelin, A. Chem. -Eur. J. 2014, 20, 6828.
(b) Moragas, T. ; Correa, A. ; Martin, R. Chem. -Eur. J. 2014, 20, 8242.
(c) Weix, D. J. Acc. Chem. Res. 2015, 48, 1767.
(d) Wang, X. ; Dai, Y. ; Gong, H. Top. Curr. Chem. 2016, 374, 43.
(a) Ye, Y. ; Zhou, Q. ; Zheng, R. ; Jiang, H. ; Chen, R. ; Zhang, Y. Appl. Organomet. Chem. 2011, 25, 331.
(b) Xia, Y. ; Chen, X. ; Qu, L. ; Sun, K. ; Xia, X. ; Fu, W. ; Chen, X. ; Yang, Y. ; Zhao, Y. ; Li, C. Asian J. Org. Chem. 2016, 5, 878.
(c) Sun, X. ; Wang, L. ; Zhang, Y. Synth. Commun. 1998, 28, 1785.
(d) Srinivas, K. ; Dubey, P. K. Synth. Commun. 2011, 41, 1584.
(e) Saikia, P. ; Laskar, D. D. ; Prajapati, D. ; Sandhu, J. S. Chem. Lett. 2001, 512.
(f) Fu, Y. ; Xu, Q. -S. ; Li, Q. -Z. ; Du, Z. ; Wang, K. -H. ; Huang, D. ; Hu, Y. Org. Biomol. Chem. 2017, 15, 2841.
Naeimi, H.; Nazifi, Z. S. Russ. Chem. Bull. 2015, 64, 1814. doi: 10.1007/s11172-015-1076-2
Compared to other additives such as NaI and TBAI, I2 exhibited better promoting effect and gave a higher yield. It is likely that I2 has better solubility than NaI and TBAI in DMA at room temperature. The exact reason for this is unclear at the moment. Besides the possible halogen exchange process, I2 might also activate the surface of manganese to promote the reaction.
Chen, J.; Liu, D.; Butt, N.; Li, C.; Fan, D.; Liu, Y.; Zhang, W. Angew. Chem., Int. Ed. 2013, 52, 11632. doi: 10.1002/anie.201306231
Pravst, I.; Zupan, M.; Stavber, S. Tetrahedron 2008, 64, 5191. doi: 10.1016/j.tet.2008.03.048
Tang, X.; Huang, L.; Xu, Y.; Yang, J.; Wu, W.; Jiang, H. Angew. Chem., Int. Ed. 2014, 53, 4205. doi: 10.1002/anie.201311217
Qian, H.; Huang, X. Synthesis 2006, 1934. doi: 10.1360/N052017-00137?slug=full%20text
Kumar, A.; Muthyala, M. K. Tetrahedron Lett. 2011, 52, 5368. doi: 10.1016/j.tetlet.2011.08.035
Rothberg, I.; Sundoro, B.; Bslanikas, G.; Kirsch, S. J. Org. Chem. 1983, 48, 4345. doi: 10.1021/jo00171a037
Weinstock, J.; Pearson, R. G.; Bordwell, F. G. J. Am. Chem. Soc. 1956, 78, 3468. doi: 10.1021/ja01595a055
Garcı́a-Ruano, J. L.; Rumbero, A. Tetrahedron:Asymmetry 1999, 10, 4427. doi: 10.1016/S0957-4166(99)00467-X
Table 1. Optimization of the reaction conditionsa
| |
||||
| Entry | Reductant | Additive | Solvent | Yieldb/% |
| 1 | Fe | none | DMA | 20 |
| 2 | Zn | none | DMA | trace |
| 3 | Mn | none | DMA | 52 |
| 4 | Mn | NaI | DMA | 58 |
| 5 | Mn | TBAI | DMA | 65 |
| 6 | Mn | I2 | DMA | 86 |
| 7 | Mn | MgCl2 | DMA | 56 |
| 8 | Mn | TMSCl | DMA | 68 |
| 9 | Mn | TFA | DMA | 61 |
| 10 | Mn | I2 | DMF | 59 |
| 11 | Mn | I2 | DMPU | 73 |
| 12 | Mn | I2 | NMP | 57 |
| 13 | Mn | I2 | DMSO | 15 |
| 14 | Mn | I2 | THF | 0 |
| 15 | Mn | I2 | CH3CN | 0 |
| a All reactions were carried out with 1a (0.20 mmol), 2a (0.30 mmol), reductant (0.60 mmol), and additive (0.020 mmol) in solvent (0.80 mL) at room temperature for 24 h. DMA=N, N-dimethylacetamide, TBAI=tetrabutylammonium iodide, TMSCl=trimethylsilyl chloride, TFA=tri-fluoroacetic acid, DMPU=1, 3-dimethylpropyleneurea, NMP=N-methyl-2-pyr-rolidone. b Isolated yield based on 1a. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Substrate scope of α-bromoketonesa
|
||
|
|
|
| |
|
|
| |
|
|
| |
|
|
| |
||
| a All reactions were carried out with 1 (0.20 mmol), 2a (0.30 mmol), Mn (0.60 mmol), and I2 (0.020 mmol) in DMA (0.80 mL) at room temperature for 24 h. Isolated yield based on 1. | ||
下载: 导出CSV
Table 3. Substrate scope of sulfonyl chloridesa
| |
|
| |
|
| |
|
| |
|
| |
|
| a All reactions were carried out with 1a (0.20 mmol), 2 (0.30 mmol), Mn (0.60 mmol), and I2 (0.020 mmol) in DMA (0.80 mL) at room temperature for 24 h. Isolated yield based on 1a. | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们