Table 1.
Comparison of CO2 adsorption capacity on different solid adsorbents.
Citation:
Zixuan Zhu, Xianjin Shi, Yongfang Rao, Yu Huang. Recent progress of MgO-based materials in CO2 adsorption and conversion: Modification methods, reaction condition, and CO2 hydrogenation[J]. Chinese Chemical Letters,
2024, 35(5): 108954.
doi:
10.1016/j.cclet.2023.108954

Recent progress of MgO-based materials in CO2 adsorption and conversion: Modification methods, reaction condition, and CO2 hydrogenation
English
Recent progress of MgO-based materials in CO2 adsorption and conversion: Modification methods, reaction condition, and CO2 hydrogenation
Abstract:
Integrated CO2 capture and conversion of (ICCC) is one of the most effective solutions to reduce anthropogenic CO2 emissions, which has attracted extensive public attention. Dual functional materials (DFMs), including adsorbent and catalyst, are the key components to achieve ICCC. Magnesium oxide (MgO) is an ideal adsorbent for ICCC, since it is characterized by high theoretical adsorption capacity, low cost, low energy consumption and extensive sources. It can also be used as DFMs in combination with the Ni catalysts. MgO not only can act as an adsorbent in DFMs but also enhance the catalytic performance of Ni. This review summarizes the advantages and modification methods of MgO as adsorbent and the influence of its adsorption conditions on the adsorption performance. Moreover, the important role of MgO in facilitating the catalytic conversion of CO2 is highlighted. Future research focuses are proposed for the development of MgO based DFMs with high adsorption capacity, high stability, conversion, and selectivity as well as low cost and energy consumption.
-
Key words:
- CO2
- / ICCC
- / MgO
- / Adsorption
- / Conversion
-
1. Introduction
Global climate change has aroused widespread concern [1,2]. Compared with the level before industrialization (~1880), global average surface temperature increases about 0.92 ℃ in 2012 [3], and is expected to increase about 6 ℃ in 2050 [4]. Scientists generally believe that anthropogenic greenhouse gases released into the atmosphere are the main cause of global climate change [5]. CO2 is the most significant components of greenhouse gas, which is the main contributor to the greenhouse effect [6]. Atmospheric CO2 concentrations reached 400 ppm in 2015, significantly higher than the pre-industrial revolution level of 280 ppm [7]. The primary source of anthropogenic CO2 is the burning of fossil fuels (coal, oil, and natural gas) [8]. Besides, fossil fuels will still be the main energy source that human beings rely on in the coming decades, so it is particularly urgent to develop effective technologies to reduce CO2 emissions [9].
Carbon capture, utilization, and storage (CCUS) is a crucial technology to reduce CO2 emission [10,11]. However, CCUS suffers from technical immaturity, excessive costs, energy consumption and risks in the transport and storage of CO2. CCUS lacks pipeline failure models during transport and storage risk assessment system [12]. In addition, the cost and energy loss incurred by CO2 transport and storage significantly increase the operating costs of CCUS [13]. The long-distance transport of CO2 can lead to dangerous accidents due to changes in external conditions or materials corrosion [13], such as major hazards to the environment, humans and ecosystems caused by CO2 leakage [14]. On the other hand, the geological storage of CO2 is highly uncertain: CO2 may leak through faults and fractures, leading to earthquakes [12]. To address these issues, researchers have proposed the integrated CO2 capture and conversion (ICCC) technology, which is the employment of in situ adsorption and conversion of CO2 to value-added product, such as methane (CH4), carbon monoxide (CO), methanol (CH3OH), cyclic carbonates, hydrocarbon and dimethyl ether [15–21]. It offers new perspectives for the reduction of CO2 emissions and the synthesis of natural gas. Especially, CO2 capture and hydrogenation have received extensive attention [22–24].
Dual functional materials (DFMs) are ideal for ICCC [25]. Developing DFMs with high adsorption capacity, high conversion, high stability, and low cost is the focus and challenge of ICCC technology. Solid adsorbents as alternative CO2 separation materials can reduce capture costs and increase CO2 adsorption [26]. Solid adsorbents can be divided into physical adsorbents (e.g., zeolite, carbon adsorbents and MOFs) and chemical adsorbents (e.g., metal oxides and hydrotalcite-like compounds) according to adsorption mechanisms [27–31]. Chemical adsorbents are more attractive due to their high selectivity and adsorption capacity [32]. Metal oxides (especially alkali metal and alkaline earth metal oxides) as chemical adsorbents have attracted much attention due to their low cost, wide availability, and high specific surface area. MgO [33], CaO [34] and Li2ZrO3 [35] have been studied and used as adsorbents for CO2. However, CaO and Li2ZrO3 is a typical high-temperature adsorbent, which is not conducive to CO2 methanation in thermodynamics. MgO achieves CO2 adsorption at moderate temperatures and has a higher theoretical adsorption capacity than CaO and Li2ZrO3. Therefore, MgO has become one of the most promising adsorbents for CO2 capture at medium temperature [35,36]. Meanwhile, MgO is also a suitable metal support for CO2 reduction catalysts. It can provide basic centers for the catalysts. These basic centers not only promote Ni reduction but also inhibits the sintering of Ni catalysts on the catalyst surface [37–39].
However, the actual adsorption capacity of MgO (0.24 mmol/g) is poor [40]. Great efforts devoted to improving the adsorption performance of MgO based adsorbents include (1) adjusting the pore size, (2) reducing the particle size, (3) dispersing MgO on the support, (4) loading metals/metal oxides, and (5) modifying MgO using molten salts [41–44]. MgO plays the role of adsorbent in DFMs. The optimum adsorption temperature of MgO coincides with the optimum temperature of the methanation reaction, thus rendering MgO an ideal adsorbent prior to CO2 methanation reaction. Combining MgO with catalysts such as Ni enables the capture and methanation of CO2 and is an effective way to reduce carbon emissions.
The potential of MgO to enhance CO2 adsorption performance and its contribution to conversion has not been described comprehensively in previous studies. Therefore, this review focuses on the significant role of MgO in CO2 adsorption and conversion. Firstly, it summarizes the advantages and limitations of MgO based materials as CO2 adsorbents. Secondly, the modification methods of MgO based adsorbents in terms of structural and chemical properties are described. Thirdly, the effect of adsorption conditions on the CO2 adsorption performance of MgO based adsorbents is also presented. Finally, the role of MgO as ICCC adsorbent in improving the catalytic performance of the catalyst and its potential as catalyst support is emphasized.
2. Advantages and limitations of MgO as CO2 adsorbent
MgO is a significant material which has been widely used in various fields due to its low cost and multifunction properties. MgO has been used as refractory protective material [45], electrode coating for solar cells [46], photocatalyst [47], and antibacterial material [48]. Recently, the application of MgO in reducing CO2 in flue gas has attracted extensive attention as a solid CO2 adsorbent [41,49–53], due to its rich sources and low cost, as well as its high theoretical CO2 adsorption capacity. Mg element are abundant on the earth, ranking the eighth in the crustal content. The most common magnesium ore minerals are dolomite (CaMg(CO3)2), magnesite (MgCO3), brucite (Mg(OH)2), carnallite (MgCl2·KCl·6H2O), brucite (MgCl2·6H2O), olivine ((Mg, Fe)2SiO4), etc. [54]. MgCO3 and CaMg(CO3)2 are the main sources for MgO [55]. In addition, as a basic oxide, MgO can act as an adsorbent to capture acid gases (e.g., CO2). The adsorption of CO2 by MgO can be categorized into physical adsorption and chemical adsorption. Physisorption of CO2 occurs on Mg2+ sites [56] at temperatures of approximately 25–40 ℃ [50]. Chemical reaction is the main pathway for CO2 capture by MgO: MgO reacts with CO2 to form MgCO3 (Eq. 1) [57]:
(1) Theoretically, 1 g MgO can adsorb 24.8 mmol of CO2 [35], which is higher than most solid adsorbents (Table 1). Although the CO2 adsorption capacity of metal-organic framework (MOF) is the highest among all adsorbents reported at present [58], MOF has the disadvantages of high cost, poor moisture resistance [59], and lower stability than chemical adsorbents [60]. In addition, compared to other metal oxides, MgO not only has higher theoretical adsorption capacity (17.8 mmol/g and 6.53 mmol/g for CaO and Li2ZrO3, respectively) (Table 1), but also has lower adsorption-desorption temperature (300–400 ℃), implying that CO2 adsorption–desorption of MgO requires lower energy consumption [35], further reducing the operating cost.
Table 1
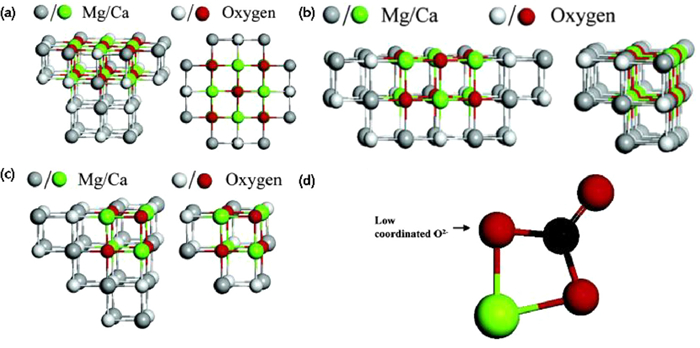
The strength of the interaction between CO2 and MgO surface is related to the surface energy of the crystalline surface, the coordination number of the surface O atoms and the ability to donate electrons [61,62]. (110), (111) and (100) are the three thermodynamically most stable surfaces of MgO. The (111) surface consists of two alternating layers of Mg and O atoms with (111)/O and (111)/Mg terminated surface. CO2 is weakly adsorbed on the (100) and (111)/O, and strongly adsorbed on the (110) surface [61,62]. CO2 chemisorption sites are low coordination O ions and oxygen vacancies on the surface of MgO [56,61,63]. O5c sites are generally more stable and inert toward CO2 [64]. O4c and O3c sites with lower coordination number than O5c, have higher reactivity toward CO2. Compared to the terraces (Fig. 1a), O4c sites are more stable at the edge (Fig. 1b) and forms monodentate carbonates. O3c sites are more reactive at the corner sites (Fig. 1c) than those at the edge and terrace sites, producing bidentate (Fig. 1d) and tridentate carbonates [65].
Figure 1
 Figure 1. CO2 adsorption model at different positions on MgO surface ((front (left) and top (right) view): (a) MgO/CaO terrace site; (b) MgO/CaO edge site; (c) MgO/CaO corner sites) and bidentate carbonate formed at corner site of O3c sites (d). Copied with permission [65]. Copyright 2005, American Chemical Society.
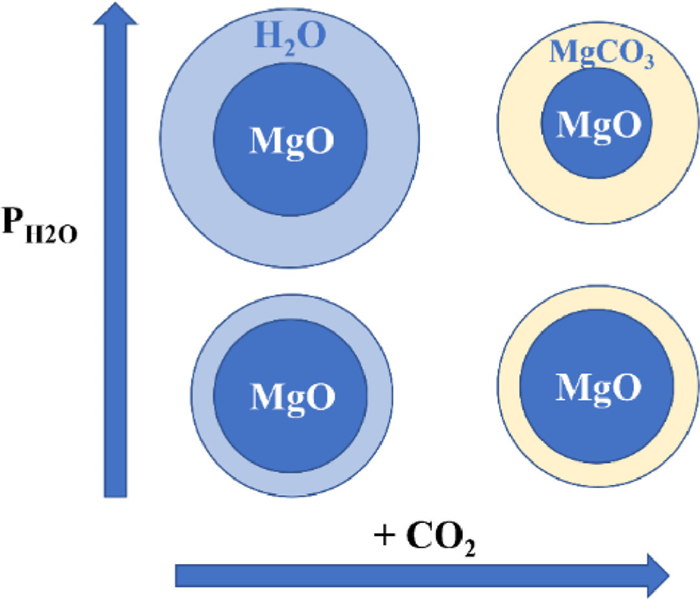
Figure 1. CO2 adsorption model at different positions on MgO surface ((front (left) and top (right) view): (a) MgO/CaO terrace site; (b) MgO/CaO edge site; (c) MgO/CaO corner sites) and bidentate carbonate formed at corner site of O3c sites (d). Copied with permission [65]. Copyright 2005, American Chemical Society.However, previous studies have shown that CO2 actual adsorption amount (0.24 mmol/g) of MgO is much less than the theoretical adsorption (24.8 mmol/g) [35,40]. This can be explained as follows: (1) High enthalpy of the reaction between MgO and CO2 results in weak adsorption, limiting the reaction rate of carbonation [40,43]; (2) Few basic sites (O2−) are exposed on the MgO surface [66]; (3) The adsorption of CO2 by MgO forms a carbonate layer on the surface of the adsorbent (Fig. 2), especially the presence of water accelerates the formation of carbonate layer [67,68]. Currently, structure modification has been applied to improve the exposure of MgO to CO2 [19–23]. It has also been demonstrated that adsorption capacity of MgO based adsorbents can be enhanced by the increase of surface alkalinity and the decrease of the energy barrier for chemical reactions [42,43,67]. Section 3 in this paper focuses on the achievement of MgO modification.
Figure 2
 Figure 2. Adsorption process of CO2 on the surface of MgO in different water vapor partial pressures. Reproduced with permission [68]. Copyright 2012, RSC Advances.
Figure 2. Adsorption process of CO2 on the surface of MgO in different water vapor partial pressures. Reproduced with permission [68]. Copyright 2012, RSC Advances.3. Modification of MgO based adsorbents
3.1 Structural modification of MgO based adsorbents
CO2 is adsorbed by MgO mainly via the interaction between CO2 and alkaline sites (O2−) [65]. The structure modification of the adsorbent can increase the number of basic sites, the specific surface area and pore diameter of the adsorbent [57]. High specific surface area is conducive to the exposure of basic sites and the adsorption of CO2 on MgO [51,66,69,70]. Table 2 shows the CO2 adsorption capacity of MgO with different specific surface area. However, the increase of specific surface area cannot guarantee the increase of adsorption capacity [71]. Pore structure has also been reported to influence the adsorption capacity of MgO. Ordered pore structure can accelerate the CO2 capture rate of adsorbent, thus improving the adsorption capacity [72].
Table 2
 Table 2. Effect of change in specific surface area and pore volume on adsorption capacity of MgO based adsorbents at different temperature.
Table 2. Effect of change in specific surface area and pore volume on adsorption capacity of MgO based adsorbents at different temperature. DownLoad:
CSV
DownLoad:
CSV

Reducing the particle size can also increase the specific surface area of the adsorbent [73], leading to the increased exposure of basic sites. MgO nanoparticles have more basic sites due to their small size, which exposes more O2− on the edges and corners compared to bulk MgO [41]. Furthermore, nano size of MgO particles was reported to inhibit the sintering of adsorbent, and thus improve the cycle stability [74]. Elvira et al. prepared MgO nano powders from CO(NH2)2 and Mg(NO3)2 (2:1) by solution-combustion and ball-milling processes [53]. Its adsorption capacity at 25 ℃ and 1 atm was 1.61 mmol/g and was 1.47 mmol/g after five adsorption–desorption cycles. However, small particle size may have negative effect on the practical application of MgO powder. Powdered adsorbents suffer from increased pressure drop or elutriation when being applied to industrial fixed or fluidized beds for CO2 separation [41].
Synthesis of porous adsorbent is another method to improve specific surface area and pore size. MgO-based porous adsorbents include porous MgO [66,69,73,75,76] and MgO dispersing on porous substrates [71,77]. Mesoporous materials as ideal porous adsorbents with pore diameters of 2–50 nm have high specific surface area, high alkalinity and ordered pore structure (Table 2). Hanif et al. synthesized MgO by ammonia precipitation, urea hydrolysis and thermal degradation methods. They found that mesoporous MgO had the highest CO2 adsorption capacity (~1.75 mmol/g) with a high specific surface area (361 cm2/g) at 200 ℃, 1 bar [76]. TiO2 [78], Al2O3 [42,79,80], CeO2 [42,81], SiO2 [82], ZrO2 [83], CuO [84] and porous carbon materials [75] have been used to support MgO [85]. Mesoporous carbon stabilized MgO is the ideal CO2 adsorbents among the above materials. Liu et al. obtained mesoporous carbon stabilized MgO nanoparticles (mPC-MgO) from MgCl2 and biomass waste [41]. TEM images showed that MgO nanoparticles were uniformly distributed on the carbon sheet. They found that the CO2 adsorption capacity of mPC-MgO exceeded 2 mmol/g at 200 ℃ (greater than other MgO-based adsorbents in Table 1). The maximum adsorption (5.45 mmol/g) was reached at 80 ℃. Moreover, mPC-MgO showed good selectivity toward CO2: the amount of CO2 being captured (5.22 mmol/g) was much higher than that of N2 (0.045 mmol/g) and O2 (0.47 mmol/g).
The morphology of adsorbents also influences the CO2 capture of MgO based adsorbents. Gao et al. prepared MgO nanosheet a with high specific surface area (327 m2/g, 0.38 cm3/g) using a thin sheet structure of hydro magnesite as a precursor [66]. The maximum adsorption capability of MgO nanosheets they prepared was 1.99 mmol/g at 60 ℃. Patchanee et al. fabricated microspheres with high surface area (227 m2/g) and high pore volume (0.79 cm3/g) using spherical magnesium ethanol as precursors [86]. The microspheres had a high adsorption capacity of 6.85 mmol/g at 25 ℃ and 0.6 MPa CO2 pressure. Doping irregular metal oxide is another method to change the morphology of adsorbent. Chen et al. studied the structure and adsorption properties of FeO–MgO [87]. They found that the core-shell structure of FeO–MgO increased the surface curvature of MgO, promoting the exposure of defective sites at the edges and corners of the crystal surface, thus providing more alkaline sites to adsorb CO2. Instantaneous adsorption-temperature programmed desorption indicated the proportion of strong alkaline sites on FeO–MgO surface increased to 91%, higher than that on MgO (28.9%).
3.2 Other modifications of MgO adsorbents
The surface oxygen ions (O2−) of MgO are the main basic sites of capturing CO2, and the coordination states of the different O2− determine the CO2 adsorption properties [65]. O2− at the edge or corner of the lattice are usually more alkaline, which react with CO2 to form monodentate, bidentate or tridentate carbonate, respectively [64,88]. More basic sites can increase the surface alkalinity of the adsorbent, thus boosting the CO2 adsorption capacity. In addition, reducing the reaction energy barrier or increasing the adsorption energy to improve the carbonation reaction rate has also become research focuses in the field of MgO modifications [43]. Metals, metal oxides and alkali metal molten salts can be used as MgO promoters to improve its CO2 adsorption capacity by increasing the surface alkalinity, lowering the reaction energy barrier, or increasing the adsorption energy.
3.2.1 Increase of surface alkalinity on MgO based adsorbents
Metal oxides such as ZnO, La2O3, CeO2, and CuO have been proved to improve the CO2 adsorption capacity of MgO based adsorbents [42,81,84,87,89–92] by increasing the alkalinity of MgO. CuO, ZnO and La2O3 alter the number of basic sites by replacing Mg2+ with metal ions, thus improving the adsorption performance. The substitution of Mg2+ (0.066 nm) by Cu2+ (0.073 nm) [84] and La3+ (0.115 nm) with larger radii than Mg2+ [92], or Zn2+ (0.060 nm) smaller than Mg2+ [93] could create structural defects, leading to the exposure of more active sites on the surface of MgO by producing an increase in the alkalinity of MgO. The remaining Cu2+ could maintain the pore structure of the adsorbent, acting as a support to disperse MgO [84].
The variation in crystalline phase could also change surface alkalinity of MgO. Vishwanath et al. synthesized mesoporous MgO–TiO2 with different TiO2 contents. They found that doping different amount of TiO2 influenced the crystalline phases of MgO and Ti oxidation state, which determined CO2 adsorption capacity of the adsorbent. MgTi2O4 with spinel structure had higher alkalinity and CO2 adsorption capacity than other crystal phases (MgTiO3 and Mg2TiO4) [89].
Previous studies indicated increasing alkalinity was a more beneficial way to improve the CO2 adsorption capacity of MgO based adsorbent than increasing the specific surface area [42,81]. The recycle stability and adsorption performance of MgO coupled by Al2O3 and CeO2 at medium temperature (200 ℃) are better than that of MgO. Al2O3 improves the adsorption performance of MgO mainly by increasing the specific surface area. CeO2 improves the adsorption capacity mainly by increasing the alkalinity of the adsorbent. Although Al2O3–MgO has a larger specific surface area and pore volume, and more homogeneous pore structure than CeO2–MgO, its CO2 adsorption capacity is still lower than that of CeO2–MgO.
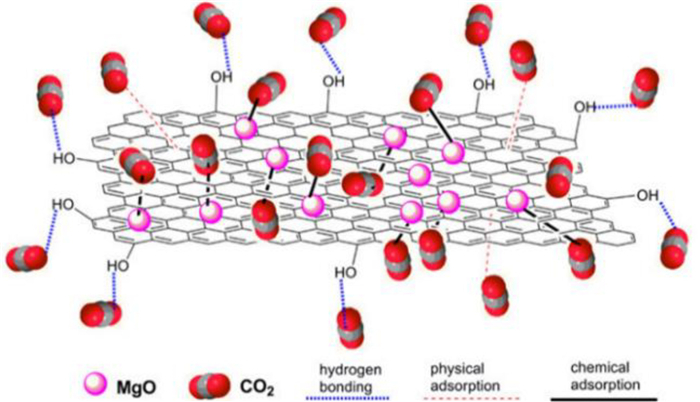
Moreover, the introduction of surface functional groups (e.g., OH) is a potential way to improve the surface properties to increase the adsorption capacity. Due to the strong electro positivity of the hydrogen atoms in the OH group, they can interact with the oxygen atoms in the CO2 to form hydrogen bonds (Fig. 3), thereby improving the CO2 adsorption of MgO adsorbents [41].
Figure 3
 Figure 3. Mechanism illustration of CO2 reaction with MgO promoted by -OH. Copied with permission [41]. Copyright 2013, American Chemical Society.
Figure 3. Mechanism illustration of CO2 reaction with MgO promoted by -OH. Copied with permission [41]. Copyright 2013, American Chemical Society.3.2.2 Promoting chemical reaction between MgO and CO2
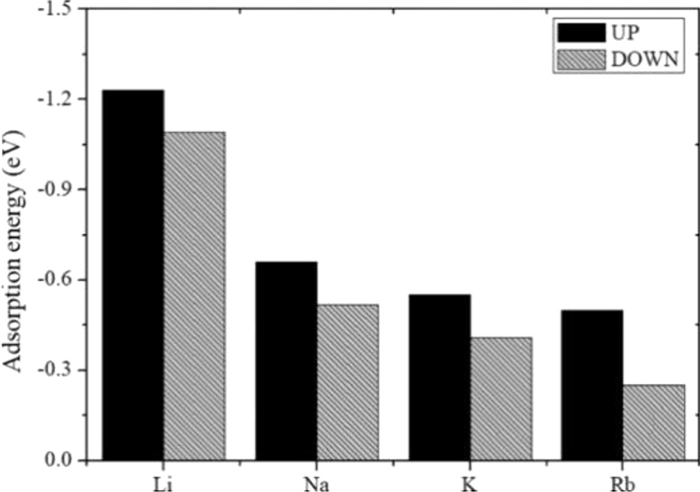
Metals can promote the adsorption capacity by increasing the adsorption energy of CO2 on MgO or decreasing the reaction energy barriers of the reaction between MgO and CO2. Alkali metal (Li, Na, K, Rb and Cs), alkaline earth metals (Be, Ca, Sr, and Ba), Fe and Al have been investigated to modify the adsorption properties of MgO [43,94–97]. Alkali and alkaline earth metals are considered to be superior promoters of MgO [43]. The increase in adsorption energy is related to the radius and electronegativity of the doped metal. In general, the smaller the radius and the higher the electronegativity, the higher adsorption energy is [96]. Thus, Li-doped MgO have the highest adsorption energy among alkali metals (Fig. 4). Exceptionally, metals with larger atomic radii (Cs and Ba) have greater adsorption energies than those with smaller radii (Na, K, Rb and Ca, Sr) due to the electrostatic interaction between Cs and Ba with neighboring Mg atoms. Considering the necessity to regenerate the adsorbent after adsorption, Li, Ca and Sr promoted MgO adsorbents are suitable as CO2 adsorbents [95].
Figure 4
 Figure 4. Adsorption energies (eV) of Li, Na, K, and Rb on a MgO–CaO (100) surface at two different configurations. Copied with permission [94]. Copyright 2019, American Chemical Society.
Figure 4. Adsorption energies (eV) of Li, Na, K, and Rb on a MgO–CaO (100) surface at two different configurations. Copied with permission [94]. Copyright 2019, American Chemical Society.Hu et al. investigated the CO2 adsorption properties of Ca/Fe/Al-doped MgO based on density flooding theory (DFT) calculations [96]. Compared to that of MgO, the stronger adsorption capability of Ca/Fe/Al-doped MgO was mainly attributed to (1) the strong electronic interaction between Ca/Fe/Al-doped MgO and CO2, (2) changes in the crystallographic parameters of MgO, and (3) the reduction of the transition state energy barrier [43]. The minimum adsorption energy of Al doped MgO (−98.15 kJ/mol) was greater than the maximum adsorption energy of Ca (−56.73 kJ/mol) and Fe (−76.70 kJ/mol) doped MgO, creating a more stable adsorption structure [87]. Therefore, Al doped had more significant effect on the CO2 adsorption capability of MgO.
They subsequently used Ce as a promoter to facilitate Al-doped MgO [43]. Compared to Al-doped MgO, CO2 adsorption energy on Ce/Al-doped MgO was higher and CO2 reaction potential was lower due to more chemically active metal atoms and less electrostatic repulsion around the adsorption sites on the surface of Ce/Al-doped MgO. Thus, Ce/Al-doped MgO has higher CO2 adsorption capacity. However, two issues should be considered for metal-doped MgO: (1) the difficult desorption due to the strong adsorption energy and (2) the blockage of pores due to metal doping and metal agglomeration [81].
Molten salt such as Alkali metal nitrates, nitrites, and carbonates (e.g., NaNO3, NaNO2, LiNO3, KNO3, KNO2, Li3PO4 and Na2CO3) can also be used to increase the CO2 adsorption of MgO [98–103]. When the ambient temperature (~300 ℃) is above the melting point of the alkali metal salts, the salts are converted from the solid phase to the liquid phase [35], facilitating the reaction between CO2 and MgO [104]. The adsorbent is then desorbed at a higher temperature (400–450 ℃) to be regenerated. It achieves CO2 capture at medium temperature and addresses the issue that the formation of carbonate layers decreases the adsorption capability of MgO-based absorbents. Therefore, alkali metal nitrates and nitrites have been used to improve the adsorption capacity of MgO more commonly than other promoters.
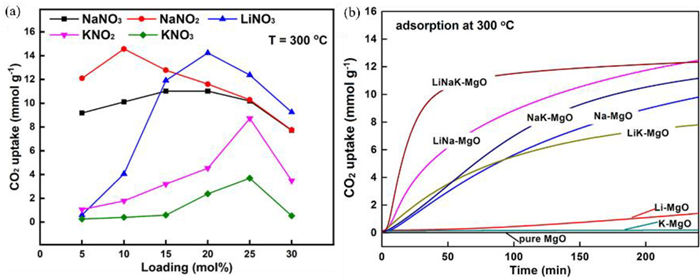
The type (unitary salt, binary salts as well as multicomponent salts) and doping amount of molten salts affects the adsorption capacity of MgO based adsorbents. Table 3 shows MgO adsorbents with different melt salts doped have various adsorption capacities. In the case of unitary salt promoted MgO, CO2 adsorption capacity of MgO doped by NaNO3, LiNO3, KNO2 and KNO3 increased with the enhancement of alkali metal nitrate loading from 5% to 20% (25%) (Fig. 5a) [104]. The adsorption capacity of NaNO2-doped MgO at 10% loading exceeded that of the other MgO materials doped by molten salts (15.7 mmol/g) [33]. In the case of binary salts promoted MgO, (NaNO3—NaNO2)/MgO had the largest CO2 uptake of 19.8 mmol/g [105]. In addition, Li–Na–K/MgO had higher CO2 adsorption capacity and adsorption rates compared to most of alkali metal nitrate doped MgO (Fig. 5b). (Li0.44K0.56)NO3]2[(Na0.5K0.5)CO3]/MgO exhibited the largest adsorption capacity among all multicomponent salt doped MgO, reaching 19.6 mmol/g [106].
Table 3
Table 3. CO2 adsorption capacities of MgO adsorbents promoted by alkali metal salts at ~300 ℃.DownLoad:
CSV

Figure 5

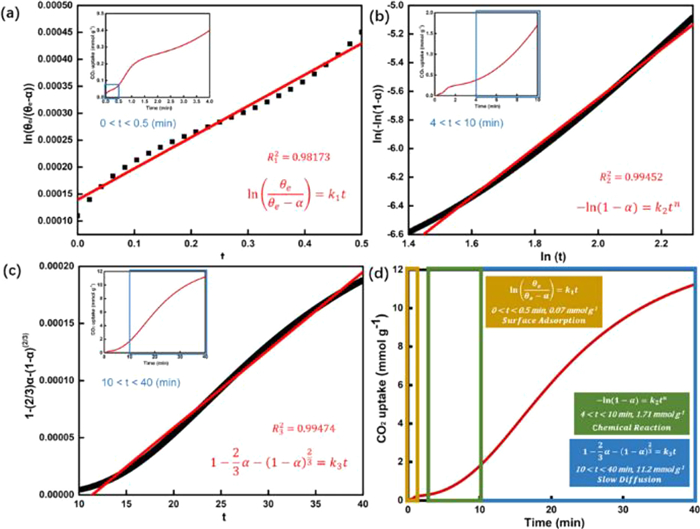
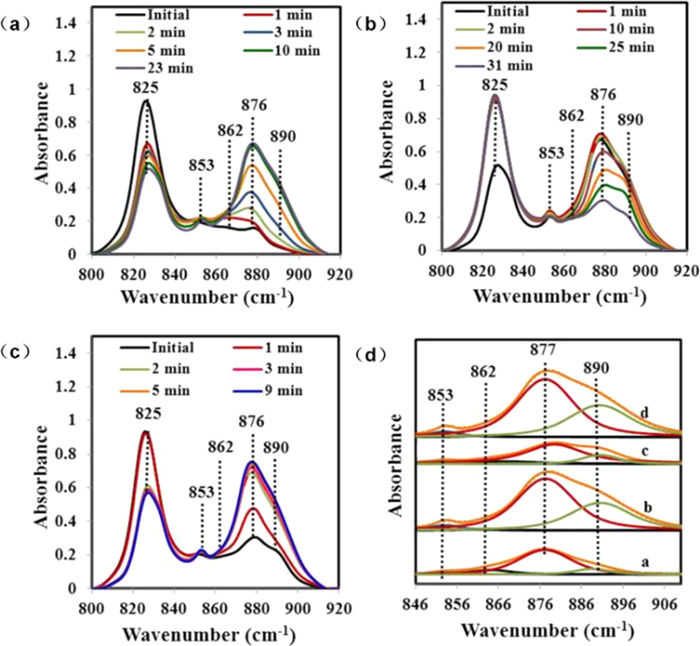
Current studies have demonstrated that doping molten salts is the most promising modification method for facilitating CO2 adsorption of MgO. Nitrate and nitrite promoted MgO has significantly enhanced the CO2 adsorption capacity, which increase from 0.24 mmol/g to a maximum of 19.8 mmol/g [105]. However, the mechanisms underlying the increased CO2 adsorption capacity of MgO promoted by molten salts remain in debate. Zhang et al. suggested that molten salts as phase transition catalysts facilitate the adsorption of gaseous CO2 on solid MgO and MgCO3 may precipitate from the initial dissolution without inhibiting the reaction between CO2 and MgO, thus improving the cyclic stability of the adsorbent. Based on DFT calculations, they found the [Mg2+-O2−] dissociation energy could be reduced by 1.771 eV with molten salt involved. Because the dissociation of the MgO ionic bond is the rate–limiting step during the CO2 adsorption process, the involvement of the molten salt increases the rate of MgCO3 production [67]. Harada and Gao et al. analyzed the reaction kinetics of CO2 adsorption using kinetic modeling [35,104]. They found that the process of CO2 adsorption on nitrate promoted MgO included three key steps: CO2 adsorption first occurred on the surface of the adsorbent with essentially linear reaction rates (Fig. 6a). Subsequently, the rate of reaction first increased and then decreased (Fig. 6b), which is attributed to the nucleation and growth of MgCO3. Due to the formation of the MgCO3 layer, the exposure of CO2 to MgO was reduced. Therefore, the reaction rate in the third stage was controlled by the diffusion of CO2 through the MgCO3 layer (Fig. 6c). In the whole adsorption process (Fig. 6d), the involvement of nitrate salt inhibited the generation of the MgCO3 layer and promoted the diffusion of CO2, accelerating the rate of the reaction in the second and third stages, thus improving the adsorption capacity of the adsorbent. Prashar et al. monitored the changes in the in-situ IR spectrum of MgO based adsorbents during multiple adsorption and desorption [107]. The nitrate (825 cm−1) peak was observed prior to the adsorption of CO2. After 1 min of reaction, the peak for nitrate decreased rapidly and the peak assigned to carbonate (862 cm−1) appeared. Subsequently, peaks for MgCO3 (853 and 890 cm−1) were observed (Fig. 7a). However, the peak at 876 cm−1 may be assigned to a lattice defect resulting from the doping of nitrate in the lattice of MgO. The nitrate peak reappeared in the desorption process (Fig. 7b). During the second adsorption, the area of the MgCO3 peak was larger than that during the first adsorption (Figs. 7c and d), implying a faster adsorption rate of CO2 due to the homogenization of nitrate in the distribution [35] or the creation of lattice defects in MgO. Gao et al. have identified the involvement of NaNO3 in CO2 adsorption by oxygen isotope labeling, including the conversion of NO3− to NO2+ and O2−, the adsorption of NO2+ on MgO, and the transfer of 18O2− from 18O−nitrate to MgO [108]. Three CO2 isotopes, C16O2 Cl6Ol8O and Cl8O2, were obtained using 18O-labelled NaNO2 (NaN l6O2 to NaN l6O218O) doped MgO (Mg18O) to adsorb CO2 for 1 h at 300 ℃. The different desorption temperatures of CO2 demonstrated the difference in the adsorption energy of CO2 on NaNO3–MgO and MgO. CO2 adsorbed by NaNO3–MgO had a higher desorption temperature, indicating the generated MgCO3 has a higher thermal stability. They also proved that NaNO3–MgO promoted the formation of [Mg2+-O2−], which increased the capacity and rate of CO2 adsorption.
Figure 6
 Figure 6. Kinetic process of nitrate promoting MgO to adsorb CO2 (d): (a) Surface adsorption process; (b) Nucleation and growth of MgCO3 phase; (c) Diffusion of CO2 in MgCO3 layer. Copied with permission [104]. Copyright 2021, American Chemical Society.
Figure 6. Kinetic process of nitrate promoting MgO to adsorb CO2 (d): (a) Surface adsorption process; (b) Nucleation and growth of MgCO3 phase; (c) Diffusion of CO2 in MgCO3 layer. Copied with permission [104]. Copyright 2021, American Chemical Society.Figure 7
 Figure 7. IR spectral changes in CO2 adsorption (a), desorption (b) and re-adsorption (c) by NaNO3-Promoted MgO and deconvolution (d) of carbonate peaks at various intervals (2 min in CO2 (curve a), 23 min in CO2 (curve b), after 30 min in He during partial desorption (curve c), and after 9 min in CO2 during re-adsorption (curve d)). Copied with permission [107]. Copyright 2016, American Chemical Society.
Figure 7. IR spectral changes in CO2 adsorption (a), desorption (b) and re-adsorption (c) by NaNO3-Promoted MgO and deconvolution (d) of carbonate peaks at various intervals (2 min in CO2 (curve a), 23 min in CO2 (curve b), after 30 min in He during partial desorption (curve c), and after 9 min in CO2 during re-adsorption (curve d)). Copied with permission [107]. Copyright 2016, American Chemical Society.However, there are some problems with alkali metal modified MgO adsorbents. During successive adsorption–desorption cycles, a reduction in the active center, blockage of the pore structure and partial depletion of the NaNO3 molten salt layer occurs on the surface of the adsorbent, leading to a gradual decrease in CO2 adsorption during the adsorption–desorption cycles [33,109]. Dong et al. suggested that the addition of Li3PO4 could provide more basic sites for the adsorbent [110], which is a possible way to solve the sintering of the adsorbent and to improve the adsorption capacity and stability.
4. Effect of adsorption conditions on the CO2 capture
The adsorption conditions such as adsorption temperature, pressure and flue gas will also affect the CO2 adsorption capacity of the adsorbents.
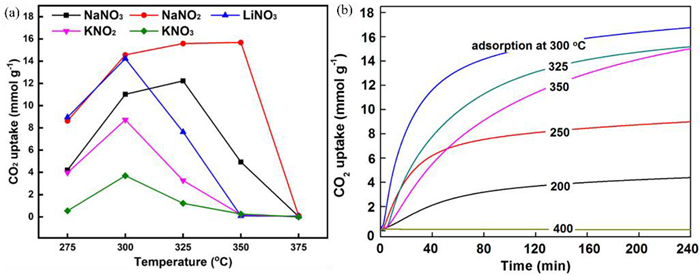
MgO can adsorb CO2 from 25 ℃ to 400 ℃ [50,73,111]. Physical adsorption is predominant at low temperature (< 40 ℃) and chemisorption occurs at higher temperatures [50]. Increasing temperature favors the reaction, but the reaction of MgO with CO2 to form MgCO3 is exothermic. The calculated Gibbs free energy suggests that the optimum adsorption temperature for MgO is 300 ℃ [104]. However, the optimum adsorption temperature varies considerably with different MgO based adsorbents, which may be related to the basic sites of the adsorbent [65,112]. Mesoporous MgO adsorbents generally exhibit high CO2 adsorption at 200 ℃ [73,76,80]. Mesoporous MgO nanoparticles with high specific surface area and high alkalinity can trap large amounts of CO2 at relatively low temperatures (< 100 ℃) [41]. The optimum adsorption temperature of molten salt modified MgO also varies with the doping of the alkali metal salt (Fig. 8a). Because the optimal adsorption temperature is related to the melting temperature of nitrate, the selection of adsorption temperature should consider the melting temperature of unitary nitrate or the eutectic point of multicomponent nitrates [49,52,108,113]. The optimum temperature of CO2 adsorption on MgO modified by binary alkali metal salts is higher than that on unitary salts modified MgO. NaNO3-promoted adsorbents have an optimum CO2 adsorption temperature of 325 ℃, while NaNO3 and NaNO2 promoted MgO adsorbents have an optimum adsorption temperature of 350 ℃. Also, NaNO3 and NaNO2 promoted MgO adsorbents show a faster CO2 uptake rate and higher adsorption stability than NaNO3-promoted adsorbents [114,115]. The optimum adsorption temperature of CO2 adsorbed by multicomponent salts is related to the ratio of molten salts. At Li: Na: K = 0.1:0.3:0.6, the adsorbent had the greatest CO2 capture capacity (16.8 mmol/g) at an adsorption temperature of 300 ℃ (Fig. 8b). At Li: Na: K = 0.3:0.18:0.52, the adsorbent had the greatest adsorption capacity (13.1 mmol/g) at 325 ℃ [103]. Adsorption temperature is also related to the partial pressure of CO2. Pressure increase facilitates CO2 adsorption [76,105].
Figure 8

The presence of gases such as H2O, O2 and SO2 in practical industrial applications can affect the adsorption capacity of MgO based adsorbents. The presence of H2O promotes the adsorption of CO2 by MgO [80,116–118]. This is probably because the OH group production from H2O dissociation on MgO surface promotes the formation of highly basic sites [49], and the adsorption energy of CO2 on MgO increases from −0.479 eV to −1.163 eV with the presence of H2O [119]. This is the same as mentioned in Section 2, where the presence of water promotes the formation of carbonates. The presence of SO2 and O2 inhibits the CO2 adsorption [102]. It is because both SO2 and CO2 are acidic gases and SO2 is more acidic than CO2. Thus, the MgO-based adsorbents prefer capturing SO2 over CO2. However, a significant increase in CO2 adsorption can be achieved in the presence of both SO2 and H2O [120]. Compared with SO2, O2 has less influence on MgO adsorption performance. It is proved that the addition of alkali metal can reduce the influence of SO2 and O2 on the adsorption performance by improving the adsorption energy, but the desorption temperature of CO2 the adsorbent also increases [97].
5. Application of MgO in CO2 conversion
Considering the problems of transportation and storage in CCUS, ICCC has received an extensive attention in recent years [15,121–124]. Among ICCC, CO2 adsorption and hydrogenation process can convert CO2 into CH4, which has increased economic benefits and aroused widespread interests [22,125,126]. The selection of CO2 adsorbent and catalyst is the key to achieve CO2 capture and conversion. As mentioned above, MgO is a suitable adsorbent for CO2 at the temperatures consistent with the optimum reaction temperature (300–400 ℃) of methanation reaction. Therefore, CO2 capture, and conversion can be carried out at the same temperature in a single reactor [22,104]. Studies have been performed with MgO as an adsorbent for DFMs achieving good adsorption and conversion performance (Table 4). Meanwhile, MgO has the potential to act as a carrier for catalysts [127]. Therefore, MgO is a suitable material for the DFMs [128–130].
Table 4
CO2 methanation converts captured CO2 to CH4 by catalytic reaction with H2. The methanation reaction equation is shown in Eq. 2 [131]:
(2) Ruthenium (Ru) and nickel (Ni) are often used as catalysts for CO2 methanation. The noble metal catalysts are highly efficient in conversion. Ru exhibits fast CO2 methanation kinetics and unique redox chemistry. However, the high cost and catalyst sintering limit the application of Ru in DFMs [132,133]. Ni catalysts with high abundance, low cost and good catalytic properties have been widely used in CO2 reduction [134]. However, Ni catalyst sintering leads to decreased specific surface area and shortened lifetimes, which reduces its catalytic ability and increases carbon deposits [135]. Ni reacts with CO at low temperatures to form nickel subcarbonyls, resulting in reduced activity [136]. In addition, Ni catalysts can suffer Ni oxidation and deactivation during the capture phase in flue gases containing large amounts of air [23]. Therefore, further studies in improving the resistance of Ni catalysts to sintering and coking as well as increasing low-temperature activity are necessary [137].
Supporting Ni on MgO is an effective way to solve the reduction of Ni activity [138,139]. As a basic additive, MgO not only increases the amount of the basic centers, favorable for CO2 activation [140], but also strengthens OH* adsorption, which is beneficial for H2O formation [38]. The CO2 conversion and CH4 selectivity of Ni/MgO was reported to be close to 90% and 100% at about 300 ℃, respectively [141]. Huang et al. investigated the pathway of CO2 methanation on Ni/MgO surfaces by means of DFT calculations. In contrast to CO2 methanation on Ni (111) surface via O-terminal hydrogenation to form HCOOH*, the C-terminal hydrogenation of CO2 on the Ni/MgO surface is more favorable than the O-terminal since electrons can be transferred from MgO to Ni nanoribbon, leading to more electrons accumulating on C atoms of CO2. Therefore, the C-terminal of the CO2 molecule has a higher electron density [38]. The combination of Ni and MgO based adsorbents is conducive to the dispersion of Ni particles and the strong metal-carrier interaction between Ni and MgO significantly improves the Ni reduction on the Ni/MgO surface [38]. Furthermore, MgO can inhibit the growth of Ni particles, thereby improving the resistance of Ni to sintering [142] and carbon deposition [143]. Furthermore, the addition of additives can further enhance CO2 adsorption and conversion performance of Ni/MgO. We could further improve CO2 methanation activity and sintering resistance through promotion the formation of monodentate species with higher H2 activity and increasing the number of stable CO2 adsorption sites [144]. Currently, Ni/MgO as a bifunctional catalyst for CO2 adsorption and methanation using alkali metal salts as promoters has obtained good performance with CO2 adsorption and CH4 yield of 6.46 mmol/g and 0.85 mmol/g at 450 ℃, respectively [145]. Moreover, Reverse water gas shift reaction was achieved with Ni/MgAl2O4 as the catalyst, showing 39.2% CO2 conversion and 100% CO selectivity at 700 ℃ [146]. This provides a new way to improve the adsorption and catalytic performance of DFMs.
6. Conclusions and prospects
MgO is an ideal medium temperature solid adsorbent for CO2. It is characterized by low cost, wide availability, and high theoretical adsorption capacity (~24.8 mmol/g). Although the actual adsorption capacity (0.24 mmol/g) is much lower than the theoretical value, it can be greatly increased by structural changes, and doping other species. Especially, the maximum adsorption capacity of MgO doped by alkali metal molten salt can reach 19.8 mmol/g. Transition metal Ni is a common catalyst for CO2 methanation with high activity and selectivity. The combination of MgO and Ni can realize in-situ CO2 capture and hydrogenation. Although the resistance of Ni to carbon deposition and sintering is weak, MgO as a basic oxide can improve the basicity of Ni, thereby increasing activity, selectivity, and stability of Ni-based catalysts. The addition of promoters can not only improve the adsorption capacity and cycle stability of the adsorbent, but also enhance the activity, selectivity, and stability of the catalyst.
Ni/MgO is an ideal material with a broad application prospect for ICCC. Furthermore, ICCC is a potential way to achieve carbon emission reduction and mitigate climate warming. Unfortunately, the current research on DFMs materials is relatively limited and further studies can be focused on:
(1) The actual adsorption capacity of MgO adsorbent differs greatly from the theoretical adsorption capacity. This is related to several factors such as preparation method, adsorption temperature and pressure, surface basic sites and CO2 concentration, etc. Therefore, the interaction of all factors should be considered to achieve CO2 capture efficiently. However, the current study considers fewer influencing factors. The next step can be based on machine learning high-throughput screening methods to efficiently optimize the existing modification methods or application conditions.
(2) The low added value of the CO2 conversion products makes it difficult to drive companies to independently select ICCC technology for large-scale applications. The next step is to change the existing active site to realize C—C coupling, such as C2H2, C2H5OH and other high value-added products conversion.
(3) Application studies under industrial conditions are lacking. For example, whether the presence of co-existing pollutants (NOx, SOx, etc.) in the flue gas will poison the existing catalysts is a direction that needs to be taken into account in future catalyst design and selection.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful to the Strategic Priority Research Program of the Chinese Academy of Sciences, China (Nos. XDA23010300 and XDA23010000), the National Natural Science Foundation of China (Nos. 51878644 and 41573138), and the Plan for "National Youth Talents" of the Organization Department of the Central Committee for financial support of this research.
-
-
[1]
C.L. Parker, P.A. Mooney, M.A. Webster, L.N. Boisvert, Nat. Commun. 13 (2022) 6514. doi: 10.1038/s41467-022-34126-7
-
[2]
C.H. Trisos, C. Merow, A.L. Pigot, Nature 580 (2020) 496–501. doi: 10.1038/s41586-020-2189-9
-
[3]
J.Y. Huang, Q.X. Li, Z.Y. Song, Sci. Total Environ. 816 (2022) 151656. doi: 10.1016/j.scitotenv.2021.151656
-
[4]
N. Mac Dowell, P.S. Fennell, N. Shah, G.C. Maitland, Nat. Clim. Change 7 (2017) 243–249. doi: 10.1038/nclimate3231
-
[5]
Y. Ou, C. Roney, J. Alsalam, et al., Nat. Commun. 12 (2021) 6245. doi: 10.1038/s41467-021-26509-z
-
[6]
S.K. Kim, J. Shin, S.I. An, et al., Nat. Climate Change 12 (2022) 834–840. doi: 10.1038/s41558-022-01452-z
-
[7]
Q. Wang, J. Luo, Z. Zhong, A. Borgna, Energy Environ. Sci. 4 (2011) 42–55. doi: 10.1039/C0EE00064G
-
[8]
M.M.F. Hasan, E.L. First, F. Boukouvala, C.A. Floudas, Comput. Chem. Eng. 81 (2015) 2–21. doi: 10.1016/j.compchemeng.2015.04.034
-
[9]
K.M.K. Yu, I. Curcic, J. Gabriel, S.C.E. Tsang, ChemSusChem 1 (2008) 893–899. doi: 10.1002/cssc.200800169
-
[10]
J. Yan, Z. Zhang, Appl. Energy 235 (2019) 1289–1299. doi: 10.1016/j.apenergy.2018.11.019
-
[11]
R.S. Norhasyima, T.M.I. Mahlia, J. CO2 Utiliz. 26 (2018) 323–335. doi: 10.1016/j.jcou.2018.05.022
-
[12]
J. Koornneef, A. Ramírez, W. Turkenburg, A. Faaij, Prog. Energy Combust. Sci. 38 (2012) 62–86. doi: 10.1016/j.pecs.2011.05.002
-
[13]
S. Chen, J. Liu, Q. Zhang, F. Teng, B.C. McLellan, Renew. Sustain. Energy Rev. 167 (2022) 112537. doi: 10.1016/j.rser.2022.112537
-
[14]
M. Hillebrand, S. Pflugmacher, A. Hahn, Inter. J. Greenh. Gas Control 55 (2016) 118–143. doi: 10.1016/j.ijggc.2016.10.014
-
[15]
I.S. Omodolor, H.O. Otor, J.A. Andonegui, B.J. Allen, A.C. Alba-Rubio, Ind. Eng. Chem. Res. 59 (2020) 17612–17631. doi: 10.1021/acs.iecr.0c02218
-
[16]
W. Zhang, R. Ping, X. Lu, et al., Chem. Eng. J. 451 (2023) 138715. doi: 10.1016/j.cej.2022.138715
-
[17]
R. Ping, C. Ma, Z. Shen, et al., Sep. Purif. Technol. 310 (2023) 123151. doi: 10.1016/j.seppur.2023.123151
-
[18]
F. Liu, X. Duan, X. Dai, et al., Chem. Eng. J. 445 (2022) 136687. doi: 10.1016/j.cej.2022.136687
-
[19]
S. Mou, T. Wu, J. Xie, et al., Adv. Mater. 31 (2019) 1903499. doi: 10.1002/adma.201903499
-
[20]
A.T. To, M.A. Arellano-Treviño, C.P. Nash, D.A. Ruddy, J. CO2 Utiliz. 66 (2022) 102261. doi: 10.1016/j.jcou.2022.102261
-
[21]
G. Bonura, S. Todaro, V. Middelkoop, et al., J. CO2 Utiliz. 70 (2023) 102458. doi: 10.1016/j.jcou.2023.102458
-
[22]
M.S. Duyar, M.A.A. Treviño, R.J. Farrauto, Appl. Catal. B: Environ. 168-169 (2015) 370–376.
-
[23]
M.S. Duyar, S.X. Wang, M.A. Arellano Treviño, R.J. Farrauto, J. CO2 Utiliz. 15 (2016) 65–71. doi: 10.1016/j.jcou.2016.05.003
-
[24]
M.A. Arellano Treviño, N. Kanani, C.W. Jeong Potter, R.J. Farrauto, Chem. Eng. J. 375 (2019) 121953. doi: 10.1016/j.cej.2019.121953
-
[25]
J. Chen, Y. Xu, P. Liao, H. Wang, H. Zhou, Carbon Capt. Sci. Technol. 4 (2022) 100052. doi: 10.1016/j.ccst.2022.100052
-
[26]
L. Jiang, A. Gonzalez Diaz, J. Ling Chin, A.P. Roskilly, A.J. Smallbone, Appl. Energy 245 (2019) 1–15. doi: 10.1016/j.apenergy.2019.04.006
-
[27]
R.M. Firdaus, A. Desforges, A. Rahman Mohamed, B. Vigolo, J. Clean. Prod. 328 (2021) 129553. doi: 10.1016/j.jclepro.2021.129553
-
[28]
Y. Belmabkhout, V. Guillerm, M. Eddaoudi, Chem. Eng. J. 296 (2016) 386–397. doi: 10.1016/j.cej.2016.03.124
-
[29]
X. Hou, L. Zhuang, B. Ma, et al., Chem. Eng. Sci. 181 (2018) 315–325. doi: 10.1016/j.ces.2018.02.015
-
[30]
C. Chen, D.W. Park, W.S. Ahn, Appl. Surf. Sci. 292 (2014) 63–67. doi: 10.1016/j.apsusc.2013.11.064
-
[31]
Y.J. Heo, S.J. Park, Energy 91 (2015) 142–150. doi: 10.1016/j.energy.2015.08.033
-
[32]
S. Wang, S. Yan, X. Ma, J. Gong, Energy Environ. Sci. 4 (2011) 3805–3819. doi: 10.1039/c1ee01116b
-
[33]
W. Liu, Y.F. Cai, M.J. Luo, Y. Yang, P. Li, Ind. Eng. Chem. Res. 61 (2022) 2882–2893. doi: 10.1021/acs.iecr.1c04615
-
[34]
H. Sun, Y. Wang, S. Xu, et al., Fuel 286 (2021) 119308. doi: 10.1016/j.fuel.2020.119308
-
[35]
T. Harada, F. Simeon, E.Z. Hamad, T.A. Hatton, Chem. Mater. 27 (2015) 1943–1949. doi: 10.1021/cm503295g
-
[36]
C.H. Huang, K.P. Chang, C.T. Yu, P.C. Chiang, C.F. Wang, Chem. Eng. J. 161 (2010) 129–135. doi: 10.1016/j.cej.2010.04.045
-
[37]
M. Yusuf, M. Beg, M. Ubaidullah, et al., Int. J. Hydrogen Energy 47 (2022) 42150–42159. doi: 10.1016/j.ijhydene.2021.08.021
-
[38]
J. Huang, X. Li, X. Wang, et al., J. CO2 Utiliz. 33 (2019) 55–63. doi: 10.1016/j.jcou.2019.04.022
-
[39]
X. Qingli, Z. Zhengdong, H. Kai, et al., Int. J. Hydrogen Energy 46 (2021) 27380–27393. doi: 10.1016/j.ijhydene.2021.06.028
-
[40]
S.J. Gregg, J.D. Ramsay, J. Chem. Soc. A: Inorg. Phys. Theor. A (1970) 2784–2787.
-
[41]
W.J. Liu, H. Jiang, K. Tian, Y.W. Ding, H.Q. Yu, Environ. Sci. Technol. 47 (2013) 9397–9403. doi: 10.1021/es401286p
-
[42]
H. Yu, X. Wang, Z. Shu, M. Fujii, C. Song, Front. Chem. Sci. Eng. 12 (2018) 83–93. doi: 10.1007/s11705-017-1691-6
-
[43]
P. Hu, S. Wang, Y. Zhuo, Sep. Purif. Technol. 287 (2022) 120518. doi: 10.1016/j.seppur.2022.120518
-
[44]
W. Gao, T. Zhou, Y. Gao, et al., J. Energy Chem. 26 (2017) 830–838. doi: 10.1016/j.jechem.2017.06.005
-
[45]
I. Fongkaew, B. Yotburut, W. Sailuam, et al., Sci. Rep. 12 (2022) 10063. doi: 10.1038/s41598-022-13949-w
-
[46]
S. Petnikota, N.K. Rotte, M.V. Reddy, V.V.S.S. Srikanth, B.V.R. Chowdari, ACS Appl. Mater. Interfaces 7 (2015) 2301–2309. doi: 10.1021/am5064712
-
[47]
Z. Liu, Z. Yin, C. Cox, et al., Sci. Adv. 2 (2016) e1501425. doi: 10.1126/sciadv.1501425
-
[48]
M.R. Bindhu, M. Umadevi, M. Kavin Micheal, M.V. Arasu, N. Abdullah Al-Dhabi, Mater. Lett. 166 (2016) 19–22. doi: 10.1016/j.matlet.2015.12.020
-
[49]
G. Bang, K.M. Kim, S. Jin, C.H. Lee, Chem. Eng. J. 433 (2022) 134607. doi: 10.1016/j.cej.2022.134607
-
[50]
H. Bekhti, H. Bouchafaa, R. Melouki, A. Travert, Y. Boucheffa, Microporous Mesoporous Mater. 294 (2020) 109866. doi: 10.1016/j.micromeso.2019.109866
-
[51]
S.W. Bian, J. Baltrusaitis, P. Galhotra, V.H. Grassian, J. Mater. Chem. 20 (2010) 8705–8710. doi: 10.1039/c0jm01261k
-
[52]
A. Dal Pozzo, A. Armutlulu, M. Rekhtina, P.M. Abdala, C.R. Müller, ACS Appl. Energy Mater. 2 (2019) 1295–1307. doi: 10.1021/acsaem.8b01852
-
[53]
G.B. Elvira, G.C. Francisco, S.M. Victor, M.L. Raul Alberto, J. Environ. Sci. 57 (2017) 418–428. doi: 10.1016/j.jes.2016.11.016
-
[54]
Y. González, A. Navarra, R.I. Jeldres, N. Toro, Hydrometallurgy 201 (2021) 105573. doi: 10.1016/j.hydromet.2021.105573
-
[55]
J. Yu, J. Qian, F. Wang, et al., Constr. Build. Mater. 253 (2020) 119147. doi: 10.1016/j.conbuildmat.2020.119147
-
[56]
E. Florez, P. Fuentealba, F. Mondragón, Catal. Today 133-135 (2008) 216–222.
-
[57]
Y.D. Ding, G. Song, X. Zhu, R. Chen, Q. Liao, RSC Adv. 5 (2015) 30929–30935. doi: 10.1039/C4RA15127E
-
[58]
A.R. Millward, O.M. Yaghi, J. Am. Chem. Soc. 127 (2005) 17998–17999. doi: 10.1021/ja0570032
-
[59]
S.Y. Lee, S.J. Park, J. Ind. Eng. Chem. 23 (2015) 1–11. doi: 10.1016/j.jiec.2014.09.001
-
[60]
H. Sharma, A. Dhir, Environ. Chem. Lett. 19 (2021) 851–873. doi: 10.1007/s10311-020-01118-2
-
[61]
H.X. Fan, T.Y. Cui, A. Rajendran, et al., Catal. Today 356 (2020) 535–543. doi: 10.1016/j.cattod.2020.03.010
-
[62]
M.A. Manae, L. Dheer, S. Rai, S. Shetty, U.V. Waghmare, Phys. Chem. Chem. Phys. 24 (2022) 1415–1423. doi: 10.1039/D1CP04152E
-
[63]
G. Pacchioni, Surf. Sci. 281 (1993) 207–219. doi: 10.1016/0039-6028(93)90869-L
-
[64]
D. Cornu, H. Guesmi, J.M. Krafft, H.L. Pernot, J. Phys. Chem. C 116 (2012) 6645–6654.
-
[65]
M.B. Jensen, L.G.M. Pettersson, O. Swang, U. Olsbye, J. Phys. Chem. B 109 (2005) 16774–16781. doi: 10.1021/jp052037h
-
[66]
W. Gao, T. Zhou, Q. Wang, Chem. Eng. J. 336 (2018) 710–720. doi: 10.1016/j.cej.2017.12.025
-
[67]
K. Zhang, X.S. Li, W.Z. Li, et al., Adv. Mater. Interf. 1 (2014) 1400030. doi: 10.1002/admi.201400030
-
[68]
J. Fagerlund, J. Highfield, R. Zevenhoven, RSC Adv. 2 (2012) 10380–10393. doi: 10.1039/c2ra21428h
-
[69]
G. Song, Y.D. Ding, X. Zhu, Q. Liao, Colloids Surf. A: Phys. Eng. Asp. 470 (2015) 39–45. doi: 10.1016/j.colsurfa.2015.01.061
-
[70]
H. Burri, R. Anjum, R.B. Gurram, et al., Korean J. Chem. Eng. 36 (2019) 1482–1488. doi: 10.1007/s11814-019-0346-3
-
[71]
Y.J. Heo, S.J. Park, Sci. Rep. 7 (2017) 5653. doi: 10.1038/s41598-017-06091-5
-
[72]
Y.D. Ding, X.X. Zhao, L. Chen, et al., Ind. Eng. Chem. Res. 60 (2021) 5310–5318. doi: 10.1021/acs.iecr.0c05462
-
[73]
V. Hiremath, R. Shavi, J.G. Seo, J. Colloid Interf. Sci. 498 (2017) 55–63. doi: 10.1016/j.jcis.2017.03.046
-
[74]
Z. Zhang, J. Li, J. Sun, et al., Ind. Eng. Chem. Res. 55 (2016) 7880–7887. doi: 10.1021/acs.iecr.5b03945
-
[75]
M. Bhagiyalakshmi, J.Y. Lee, H.T. Jang, Int. J. Greenh. Gas Control 4 (2010) 51–56. doi: 10.1016/j.ijggc.2009.08.001
-
[76]
A. Hanif, S. Dasgupta, A. Nanoti, Ind. Eng. Chem. Res. 55 (2016) 8070–8078. doi: 10.1021/acs.iecr.6b00647
-
[77]
A. Hanif, M. Sun, Z. Tao, et al., ACS Appl. Nano Mater. 2 (2019) 6565–6574. doi: 10.1021/acsanm.9b01481
-
[78]
H. Jeon, Y.J. Min, S.H. Ahn, et al., Colloids Surf. A: Physicochem. Eng. Asp. 414 (2012) 75–81. doi: 10.1016/j.colsurfa.2012.08.009
-
[79]
V. Hiremath, B.T. Shiferraw, J.G. Seo, J. CO2 Utiliz. 42 (2020) 101294. doi: 10.1016/j.jcou.2020.101294
-
[80]
K.K. Han, Y. Zhou, Y. Chun, J.H. Zhu, J. Hazard. Mater. 203 (2012) 341–347.
-
[81]
S. Jin, G. Bang, L. Liu, C.H. Lee, Microporous Mesoporous Mater. 288 (2019) 109587. doi: 10.1016/j.micromeso.2019.109587
-
[82]
A. Zukal, J. Pastva, J. Cejka, Microporous Mesoporous Mater. 167 (2013) 44–50. doi: 10.1016/j.micromeso.2012.05.026
-
[83]
J.S. Kwak, K.Y. Kim, K.R. Oh, Y.U. Kwon, Int. J. Greenh. Gas Control 81 (2019) 38–43. doi: 10.1016/j.ijggc.2018.12.010
-
[84]
Y.Y. Li, X.Y.M. Dong, X.D. Sun, Y. Wang, J.H. Zhu, ACS Appl. Mater. Interf. 8 (2016) 30193–30204. doi: 10.1021/acsami.6b09927
-
[85]
Y. Hu, Y. Guo, J. Sun, H. Li, W. Liu, J. Mater. Chem. A 7 (2019) 20103–20120. doi: 10.1039/C9TA06930E
-
[86]
C. Patchanee, L.T.T. Mai, T. Ikeda, M. Priyank, J. CO2 Utiliz. 51 (2021) 101652. doi: 10.1016/j.jcou.2021.101652
-
[87]
J.L. Chen, X.Y.M. Dong, C.L. Shi, et al., Clean-Soil Air Water 47 (2019) 1800447. doi: 10.1002/clen.201800447
-
[88]
N.N.A.H. Meis, J.H. Bitter, K.P. de Jong, Ind. Eng. Chem. Res. 49 (2010) 1229–1235. doi: 10.1021/ie901114d
-
[89]
V. Hiremath, R. Shavi, J.G. Seo, Chem. Eng. J. 308 (2017) 177–183. doi: 10.1016/j.cej.2016.09.052
-
[90]
W. Ueda, T. Yokoyama, Y. Morooka, T. Ikawa, Chem. Lett. 14 (1985) 1059–1062. doi: 10.1246/cl.1985.1059
-
[91]
P.Q. Lan, S.F. Wu, Chem. Eng. Technol. 37 (2014) 580–586. doi: 10.1002/ceat.201300709
-
[92]
H. Liu, K. Wang, X. Cao, J. Su, Z. Gu, RSC Adv. 11 (2021) 12532–12542. doi: 10.1039/D1RA00176K
-
[93]
Y.Y. Li, M.M. Wan, X.D. Sun, et al., J. Mater. Chem. A 3 (2015) 18535–18545. doi: 10.1039/C5TA04309C
-
[94]
J.M. Jang, S.G. Kang, ACS Sustain. Chem. Eng. 7 (2019) 16979–16984. doi: 10.1021/acssuschemeng.9b01223
-
[95]
K. Kim, J.W. Han, K.S. Lee, W.B. Lee, Phys. Chem. Chem. Phys. 16 (2014) 24818–24823. doi: 10.1039/C4CP03809F
-
[96]
P. Hu, S. Wang, Y. Zhuo, Sep. Purif. Technol. 276 (2021) 119323. doi: 10.1016/j.seppur.2021.119323
-
[97]
P. Hu, S. Wang, Y. Zhuo, Sep. Purif. Technol. 284 (2022) 120253. doi: 10.1016/j.seppur.2021.120253
-
[98]
C.H. Lee, H.J. Kwon, H.C. Lee, et al., Chem. Eng. J. 294 (2016) 439–446. doi: 10.1016/j.cej.2016.03.003
-
[99]
W. Dong, X.P. Chen, F. Yu, Y. Wu, Energy Fuels 29 (2015) 968–973. doi: 10.1021/ef502400s
-
[100]
A.T. Vu, Y. h. Park, P.R. Jeon, C.H. Lee, Chem. Eng. J. 258 (2014) 254–264. doi: 10.1016/j.cej.2014.07.088
-
[101]
S.C. Lee, S.H. Cha, Y.M. Kwon, et al., Korean J. Chem. Eng. 33 (2016) 3448–3455. doi: 10.1007/s11814-016-0185-4
-
[102]
J. Wang, M. Li, P. Lu, P. Ning, Q. Wang, Chem. Eng. J. 392 (2020) 123752. doi: 10.1016/j.cej.2019.123752
-
[103]
Y.Q. Qiao, J.Y. Wang, Y. Zhang, et al., Ind. Eng. Chem. Res. 56 (2017) 1509–1517. doi: 10.1021/acs.iecr.6b04793
-
[104]
W. Gao, M.A. Vasiliades, C.M. Damaskinos, et al., Environ. Sci. Technol. 55 (2021) 4513–4521. doi: 10.1021/acs.est.0c08731
-
[105]
X. Zhao, G.Z. Ji, W. Liu, et al., Chem. Eng. J. 332 (2018) 216–226. doi: 10.1016/j.cej.2017.09.068
-
[106]
J. Ding, C. Yu, J.F. Lu, et al., Appl. Energy. 263 (2020) 114681. doi: 10.1016/j.apenergy.2020.114681
-
[107]
A.K. Prashar, H. Seo, W.C. Choi, et al., Energy Fuels 30 (2016) 3298–3305. doi: 10.1021/acs.energyfuels.5b02909
-
[108]
W. Gao, J. Xiao, Q. Wang, et al., Adv. Mater. 34 (2022) 2106677. doi: 10.1002/adma.202106677
-
[109]
Y.H. Zheng, J.Y. Wu, L.J. Zhang, et al., Chem. Eng. J. 450 (2022) 137944. doi: 10.1016/j.cej.2022.137944
-
[110]
H. Dong, H.J. Cui, Z.M. Zhou, Chem. Eng. J. 442 (2022) 136133. doi: 10.1016/j.cej.2022.136133
-
[111]
W. Gao, T. Zhou, Y. Gao, Q. Wang, W. Lin, Energy Fuels 33 (2019) 1704–1712. doi: 10.1021/acs.energyfuels.8b02749
-
[112]
C.D. Daub, G.N. Patey, D.B. Jack, A.K. Sallabi, J. Chem. Phys. 124 (2006) 114706. doi: 10.1063/1.2171277
-
[113]
L. Zhang, Y. Zheng, Y. Guo, et al., ACS Appl. Energy Mater. 4 (2021) 9513–9524. doi: 10.1021/acsaem.1c01681
-
[114]
T. Harada, T.A. Hatton, Chem. Mater. 27 (2015) 8153–8161. doi: 10.1021/acs.chemmater.5b03904
-
[115]
K. Wang, Y.W. Zhao, P.T. Clough, P.F. Zhao, E.J. Anthony, Chem. Eng. J. 372 (2019) 886–895. doi: 10.1016/j.cej.2019.04.080
-
[116]
Y.D. Ding, G. Song, Q. Liao, X. Zhu, R. Chen, Energy 112 (2016) 101–110. doi: 10.1016/j.energy.2016.06.064
-
[117]
R.V. Siriwardane, R.W. Stevens Jr., Ind. Eng. Chem. Res. 48 (2009) 2135–2141. doi: 10.1021/ie8011598
-
[118]
N. Yang, P. Ning, K. Li, J. Wang, J. Taiwan Inst. Chem. Eng. 86 (2018) 73–80. doi: 10.1016/j.jtice.2018.02.006
-
[119]
S. Wu, B.T. Tan, H.L. Senevirathna, P. Wu, Appl. Surf. Sci. 562 (2021) 150187. doi: 10.1016/j.apsusc.2021.150187
-
[120]
A. Czyżewski, J. Kapica, D. Moszyński, R. Pietrzak, J. Przepiórski, Chem. Eng. J. 226 (2013) 348–356. doi: 10.1016/j.cej.2013.04.061
-
[121]
S. Sun, H. Sun, P.T. Williams, C. Wu, Sustain. Energy Fuels 5 (2021) 4546–4559. doi: 10.1039/D1SE00797A
-
[122]
M. Liu, P. Zhao, W. Zhang, et al., Fuel 315 (2022) 123230. doi: 10.1016/j.fuel.2022.123230
-
[123]
J. Liu, C. Chen, K. Zhang, L. Zhang, Chin. Chem. Lett. 32 (2021) 649–659. doi: 10.1016/j.cclet.2020.07.040
-
[124]
M. Liu, C. Ma, X. Cheng, et al., Sep. Purif. Technol. 317 (2023) 123937. doi: 10.1016/j.seppur.2023.123937
-
[125]
X. Shi, L.N.Y. Cao, M. Chen, Y. Huang, Chin. Chem. Lett. 33 (2022) 5023–5029. doi: 10.1016/j.cclet.2022.01.066
-
[126]
X.J. Shi, Y. Huang, Y.A. Bo, et al., Angew. Chem. Int. Ed. 61 (2022) e202203063. doi: 10.1002/anie.202203063
-
[127]
Y. Varun, I. Sreedhar, S.A. Singh, Int. J. Hydrogen Energy 45 (2020) 28716–28731. doi: 10.1016/j.ijhydene.2020.07.212
-
[128]
P. Huang, Y.F. Guo, G.D. Wang, et al., Energy Fuels 35 (2021) 20185–20196. doi: 10.1021/acs.energyfuels.1c03008
-
[129]
H.M. Sun, Y. Zhang, S.L. Guan, J. Huang, C.F. Wu, J. CO2 Utiliz. 38 (2020) 262–272. doi: 10.1016/j.jcou.2020.02.001
-
[130]
S.J. Park, M.P. Bukhovko, C.W. Jones, Chem. Eng. J. 420 (2021) 130369. doi: 10.1016/j.cej.2021.130369
-
[131]
T.A. Le, M.S. Kim, S.H. Lee, T.W. Kim, E.D. Park, Catal. Today 293-294 (2017) 89–96.
-
[132]
C.J. Potter, A. Porta, R. Matarrese, et al., Appl. Catal. B: Environ. 310 (2022) 121294. doi: 10.1016/j.apcatb.2022.121294
-
[133]
M.A. Arellano Treviño, Z.Y. He, M.C. Libby, R.J. Farrauto, J. CO2 Utiliz. 31 (2019) 143–151. doi: 10.1016/j.jcou.2019.03.009
-
[134]
W. Wang, S. Wang, X. Ma, J. Gong, Chem. Soc. Rev. 40 (2011) 3703–3727. doi: 10.1039/c1cs15008a
-
[135]
C.H. Tan, S. Nomanbhay, A.H. Shamsuddin, et al., Front. Energy Res. 9 (2022) 795423. doi: 10.3389/fenrg.2021.795423
-
[136]
J. Liu, C. Li, F. Wang, et al., Catal. Sci. Technol. 3 (2013) 2627–2633. doi: 10.1039/c3cy00355h
-
[137]
S. Natesakhawat, R.B. Watson, X. Wang, U.S. Ozkan, J. Catal. 234 (2005) 496–508. doi: 10.1016/j.jcat.2005.07.014
-
[138]
M.W. Tan, X.G. Wang, X.X. Wang, et al., J. Catal. 329 (2015) 151–166. doi: 10.1016/j.jcat.2015.05.011
-
[139]
D. Hu, J. Gao, Y. Ping, et al., Ind. Eng. Chem. Res. 51 (2012) 4875–4886. doi: 10.1021/ie300049f
-
[140]
L. Xu, F. Wang, M. Chen, et al., RSC Adv. 7 (2017) 18199–18210. doi: 10.1039/C7RA01673E
-
[141]
A.I. Tsiotsias, N.D. Charisiou, I.V. Yentekakis, M.A. Goula, Catalysts 10 (2020) 812. doi: 10.3390/catal10070812
-
[142]
J.J. Tan, J.M. Wang, Z.Y. Zhang, et al., Appl. Surf. Sci. 481 (2019) 1538–1548. doi: 10.1016/j.apsusc.2019.03.217
-
[143]
C.J. Liu, J.Y. Ye, J.J. Jiang, Y.X. Pan, ChemCatChem 3 (2011) 529–541. doi: 10.1002/cctc.201000358
-
[144]
Y. Yan, Y. Dai, H. He, Y. Yu, Y. Yang, Appl. Catal. B: Environ. 196 (2016) 108–116. doi: 10.1016/j.apcatb.2016.05.016
-
[145]
P. Huang, J. Chu, J. Fu, et al., Chem. Eng. J. 467 (2023) 143431. doi: 10.1016/j.cej.2023.143431
-
[146]
A. Ranjbar, S.F. Aghamiri, A. Irankhah, Int. J. Hydrogen Energy 48 (2023) 19115–19125. doi: 10.1016/j.ijhydene.2023.01.102
-
[1]
-
Figure 1 CO2 adsorption model at different positions on MgO surface ((front (left) and top (right) view): (a) MgO/CaO terrace site; (b) MgO/CaO edge site; (c) MgO/CaO corner sites) and bidentate carbonate formed at corner site of O3c sites (d). Copied with permission [65]. Copyright 2005, American Chemical Society.
Figure 2 Adsorption process of CO2 on the surface of MgO in different water vapor partial pressures. Reproduced with permission [68]. Copyright 2012, RSC Advances.
Figure 3 Mechanism illustration of CO2 reaction with MgO promoted by -OH. Copied with permission [41]. Copyright 2013, American Chemical Society.
Figure 4 Adsorption energies (eV) of Li, Na, K, and Rb on a MgO–CaO (100) surface at two different configurations. Copied with permission [94]. Copyright 2019, American Chemical Society.
Figure 6 Kinetic process of nitrate promoting MgO to adsorb CO2 (d): (a) Surface adsorption process; (b) Nucleation and growth of MgCO3 phase; (c) Diffusion of CO2 in MgCO3 layer. Copied with permission [104]. Copyright 2021, American Chemical Society.
Figure 7 IR spectral changes in CO2 adsorption (a), desorption (b) and re-adsorption (c) by NaNO3-Promoted MgO and deconvolution (d) of carbonate peaks at various intervals (2 min in CO2 (curve a), 23 min in CO2 (curve b), after 30 min in He during partial desorption (curve c), and after 9 min in CO2 during re-adsorption (curve d)). Copied with permission [107]. Copyright 2016, American Chemical Society.
Table 2. Effect of change in specific surface area and pore volume on adsorption capacity of MgO based adsorbents at different temperature.
 下载: 导出CSV
下载: 导出CSV
Table 3. CO2 adsorption capacities of MgO adsorbents promoted by alkali metal salts at ~300 ℃.
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 5
- 文章访问数: 1609
- HTML全文浏览量: 54
