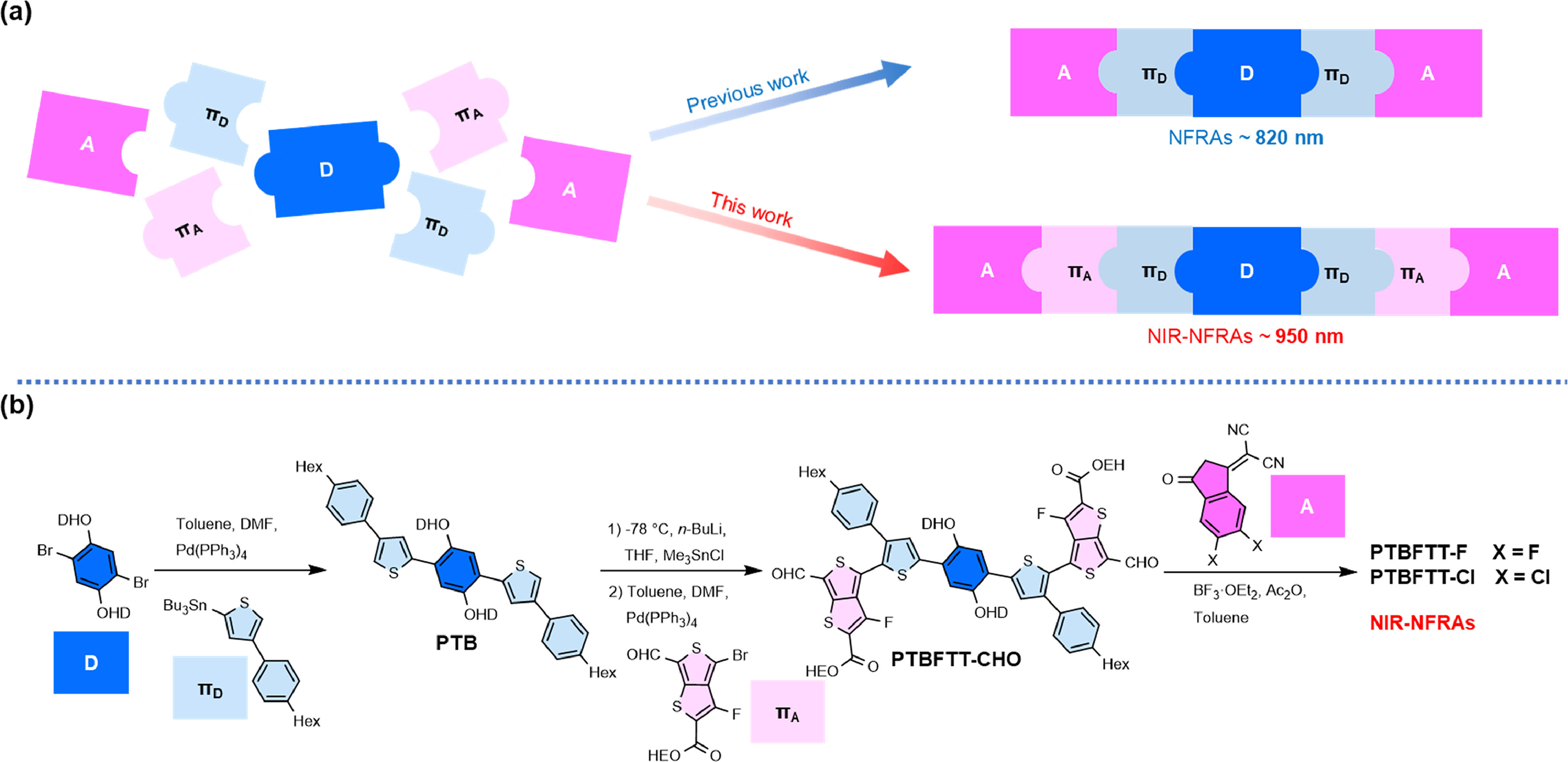
Scheme 1.
(a) Design and (b) synthetic route for NIR-NFRAs.
Near-infrared non-fused electron acceptors for efficient organic photovoltaics
Wenxiang Ma , Xinyu He , Tianyi Chen , De-Li Ma , Hongzheng Chen , Chang-Zhi Li

Organic photovoltaics (OPVs) have gained the widespread attentions [1–5], and recently achieved the power conversion efficiencies (PCEs) of over 19% in single-junction devices [6–11]. OPVs, especially semitransparent ones have great potentials to be deployed as building integrated photovoltaic (BIPV) [12–15], that requires developing narrow-bandgap photovoltaic materials with strong near-infrared (NIR) absorption [16,17]. Notably, high-performance narrow-bandgap electron acceptors, such as IEICO, IFIC-i-4F, COi8DFIC, ATT-9, FC8–2F. [18–26], share one common structural feature consisting of multiple fused-ring backbones as electron donating cores [3,22,27–30], which require constructing multiple covalent-bonds to interlock the adjunct aromatic units. These fused-ring backbones are helpful to reinforce the structural planarity and rigidity, facilitating the π-electron delocalization within the fused-ring electron acceptors (FREAs), whereas facing challenges of high synthetic complexity [31–33]. In this regard, research efforts have been devoted to explore electron acceptors with non-fused backbones, as non-fused ring electron acceptors (NFRAs), wherein the non-covalent interactions were employed to mediate the planarity and rigidity of NFRAs with the reduced synthetic complexities [4,32,34–46]. Some NFRAs, such as PTIC and PTB4Cl, can be synthesized through two-step modular reaction, meanwhile exhibiting the desirable performance and stabilities [37,39,42]. Recently, a series of tetra-thiophene and thieno[3,2-b]thiophene-based NFRAs, such as A4T-32, 4T-2F2Cl, BM-2F and L2, have been obtained, reaching the impressive PCEs of over 16% in NFRA-based OPVs [33,47–49], Nevertheless, the reported NFRAs are majorly centering their photo responses around 800 nm, or up to 900 nm of solar spectra [37,39,42,47,49,50]. Therefore, narrow-bandgap NFRAs are still needed to further red-shift the photo-response spectra, yet rarely explored [24,51–53]. To facilitate the development of OPVs with the reduced material costs, it should be desirable to explore near-infrared acceptors with excellent cost-effective features.
In this work, two near-infrared non-fused ring acceptors (NIR NFRAs), PTBFTT-F and PTBFTT-Cl are developed through the design of A-πA-πD-D-πD-πA-A non-fused structures, wherein the electron deficient π-bridge (πA) of fluorothieno[3,4-b]thiophene (FTT) is introduced to conjugate the halogenated 2-(3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile terminals with electron rich non-fused cores. Possessing multiple intramolecular noncovalent interactions within structures, both PTBFTT-F and PTBFTT-Cl retained the structural planarity and effective intramolecular charge transfer (ICT), extending strong NIR photon absorption up to 950 nm. When pairing with polymer electron donor PBDB-T, OPVs based on PTBFTT-Cl exhibited superior PCEs of 10.26% to those with PTBFTT-F acceptor (PCE of 8.57%). It is ascribed to PTBFTT-Cl, possessing the enlarged π-surface area over PTBFTT-F from the difference of halogen atom radius, promoted the molecular stacking and ordering in solid films, which is beneficial to improve the exciton and charge dynamics of the corresponding OPVs. In addition, non-fused PTBFTT-Cl exhibits the improved photochemical stabilities under one-sun equivalent irradiation in ambient (degradation rate: 2.33 × 10−3 h-1), outperforming that of NIR fused-ring acceptors. Overall, this work provides a new design strategy of simple and effective near-infrared electron acceptors with desirable practical perspective.
Two new NIR-NFRAs, PTBFTT-F and PTBFTT-Cl, have been developed via four-step reactions (Scheme 1). The commercially available 1,4-dibromo-2,5-bis((2-hexyldecyl)oxy)benzene was employed as the starting material, to couple with tributyl(4-(4-hexylphenyl)thiophen-2-yl)stannane via palladium catalyzed Stille cross-coupling reaction, affording PTB. Subsequent organostannic intermediate was prepared to couple with 2-ethylhexyl 4‑bromo-3-fluoro-6-formylthieno[3,4-b]thiophene-2-carboxylate, yielding PTBFTT-CHO. Finally, the PTBFTT-F and PTBFTT-Cl were synthesized with the yield of over 95% by Knoevenagel condensation between PTBFTT-CHO and 2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (DFIC) or 2-(5,6-dichloro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (DClIC), respectively. Further details on the synthetic procedure and structural characterization, such as proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) and matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF, Fig. S1), can be found in Supporting information. To understand the structural conformation and molecular orbitals of these new acceptors, we conducted the density functional theory (DFT) calculations based on the B3LYP/6–31G(d) level. As shown in Fig. 1 and Fig. S2 (Supporting information), there present multiple non-covalent interactions to mediate the planarity and rigidity of these non-fused acceptors. The hydrogen bonding interaction (O…H) between alkyloxybenzene and thiophene, as well as electrostatic interactions of "O…S" and "F…S", which have been confirmed in previous works [34,37,39,54–56]. The calculated results also revealed that the intramolecular distances of dintra,O-H, dintra,F-S and dintra,O-S are less than the sum of the van der Waals radii (rw, O-H = 2.50 Å, rw, F-S = 3.20 Å, rw, O-S = 3.25 Å) (Table S1 in Supporting information). Theoretical calculations on the carbon-carbon single bond torsional energy scans were performed and revealed that the central the center single bond between alkyloxy benzene and thiophene (P-TB) holds the relatively low rotation barrier, comparing to other single bonds in molecules (Fig. S3 in Supporting information). Simulated electrostatic potentials (ESP) displayed that PTBFTT-Cl possesses the enlarged π-surface area over that of PTBFTT-F, due to chlorine providing p-π conjugation with larger halogen atom radius over that of fluorine.
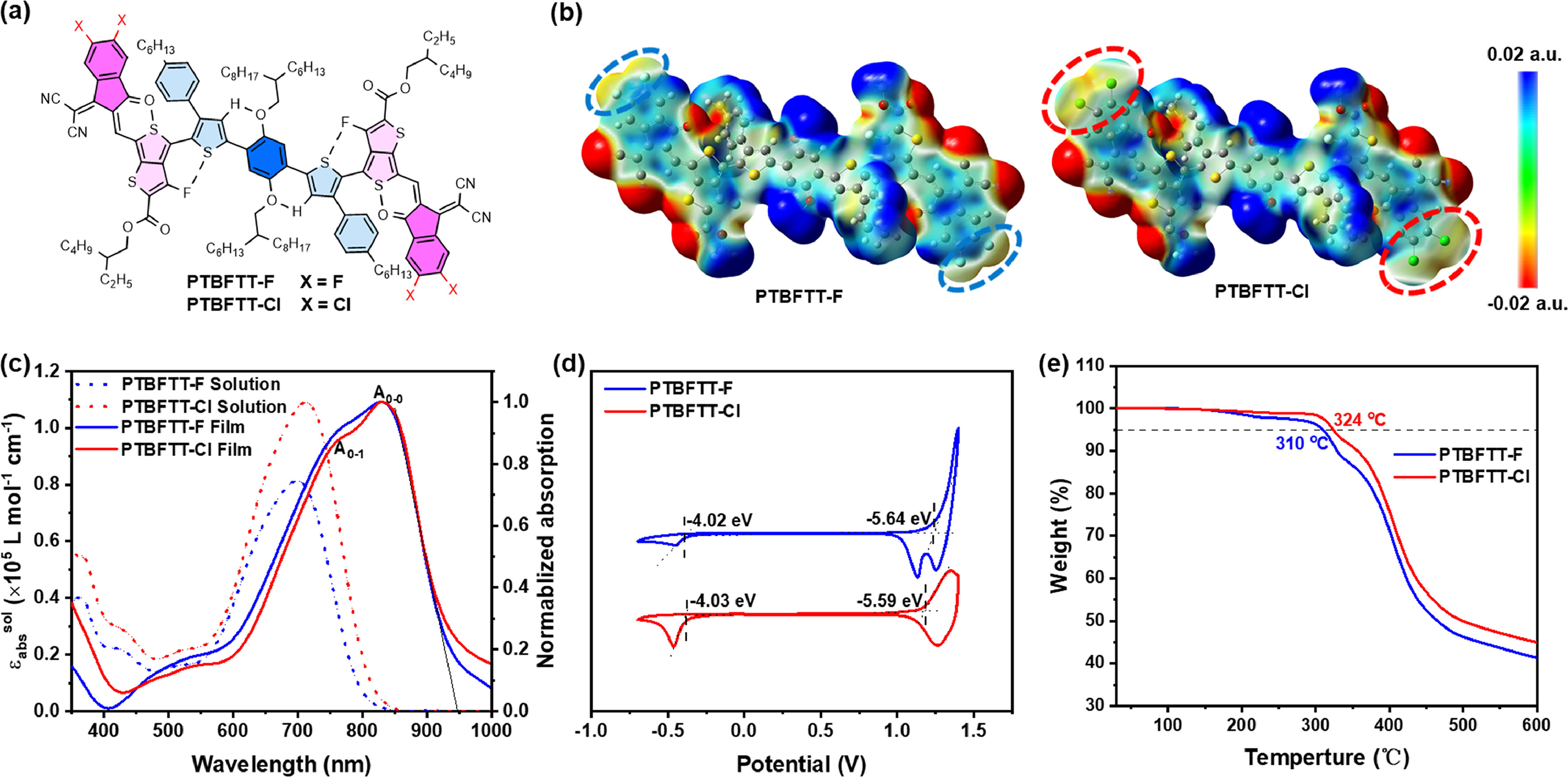
The UV–vis absorption spectra of PTBFTT-F and PTBFTT-Cl in chloroform (CF) solution and solid films are shown in Fig. 1c, and related parameters are summarized in Table 1. In CF solution, PTBFTT-Cl displays a higher molar extinction coefficient (ε) of 1.09 × 105 L mol−1 cm−1, compared to that of PTBFTT-F (8.1 × 104 L mol−1 cm−1). The maximum absorption peak (λabs, maxsol) of PTBFTT-F is located at 700 nm in solution, while that for PTBFTT-Cl takes bathochromic shift to 714 nm. For neat solid films, both the maximum absorption peaks (λabs, maxfilm) show significant bathochromic shift with 129 and 116 nm for PTBFTT-F and PTBFTT-Cl, respectively. Both PTBFTT-F and PTBFTT-Cl possess almost same absorption edge of 950 nm with narrow optical bandgaps of 1.31 eV. In addition, it is observed that PTBFTT-Cl neat films exhibit the reduced full-width at half-maximum and enhanced A0–0/A0–1 ratios of absorption spectra [57], over those of PTBFTT-F films, indicating that PTBFTT-Cl tends to form the more ordered molecular stacking in the solid. It is consistent with the results of morphology measurements (discussed hereafter).
As shown in Fig. 1d, the electrochemical cyclic voltammetry (CV) measurements were used to estimate the energy levels of PTBFTT-F and PTBFTT-Cl, showing the highest occupied molecular orbital (HOMO) levels and the lowest unoccupied molecular orbital (LUMO) levels (−5.64/−4.02 eV for PTBFTT-F, and −5.59/−4.03 eV for PTBFTT-Cl, respectively). Thermal properties were also investigated by the thermogravimetric analysis (TGA) measurements. The thermal decomposition temperatures (Td) (5% weight loss) are 310 ℃ for PTBFTT-F and 324 ℃ for PTBFTT-Cl (Fig. 1e), revealing that both acceptors possess good thermal stabilities.
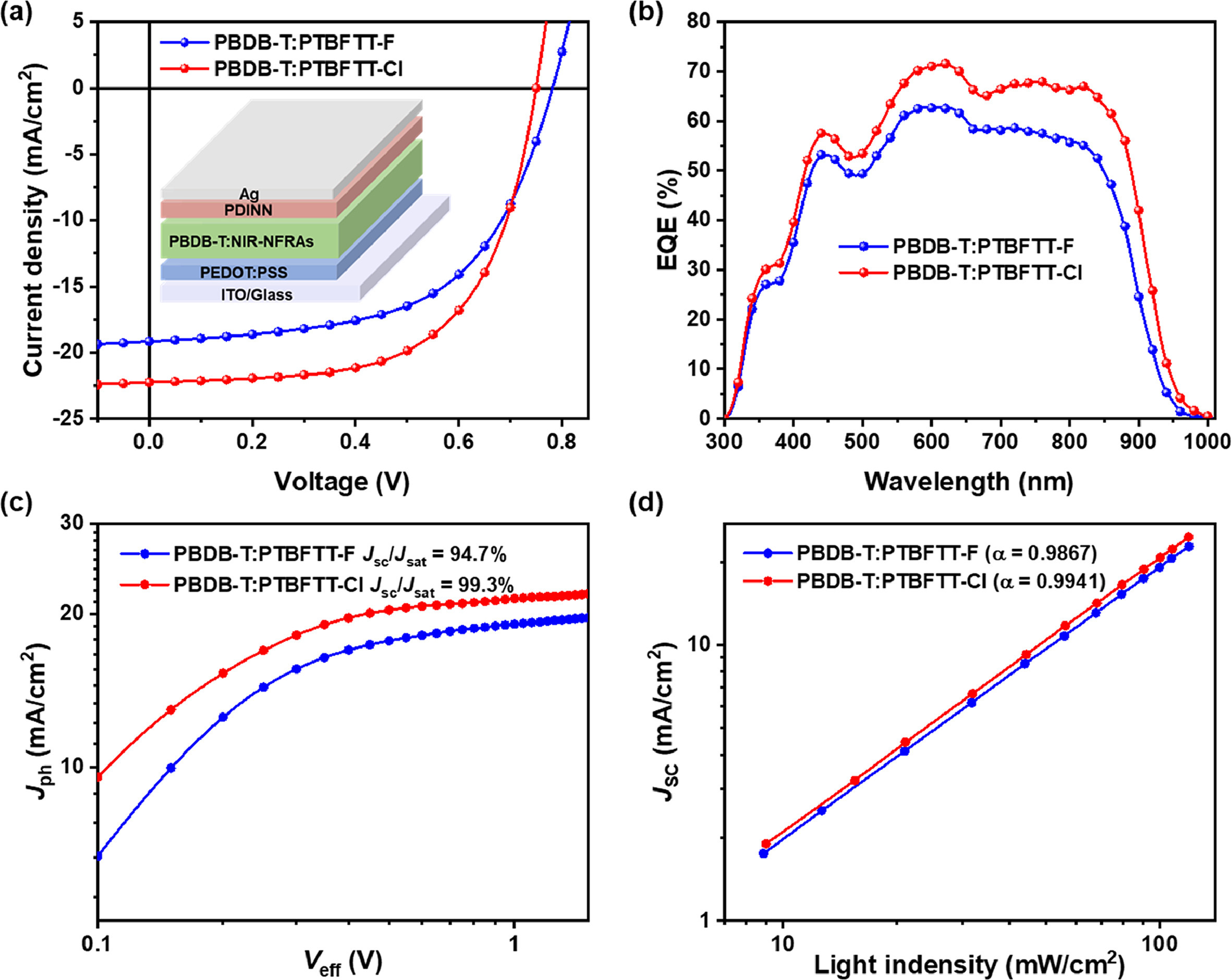
Considering the optoelectronic properties of PTBFTT-F and PTBFTT-Cl, polymer electron donor PBDB-T (Fig. S4 in Supporting information) was chosen to fabricate organic photovoltaics with the structure of indium tin oxide (ITO)/poly(3,4-ethylene dioxythiophene): poly(styrene sulfonate) (PEDOT: PSS)/(PBDB-T: NIR-NFRAs)/PDINN/Ag. The current density-voltage (J-V) curves of such OPVs are displayed in Fig. 2a and their photovoltaic parameters are summarized in Table 2. Interestingly, OPVs based on PTBFTT-Cl exhibited superior PCE of 10.26% compared to those of PTBFTT-F with PCE of 8.57%. The open-circuit voltage (Voc) of 0.78 for OPVs based on PTBFTT-F and 0.75 V for those of PTBFTT-Cl. The distinct difference of the photovoltaic performance between PTBFTT-F and PTBFTT-Cl exists in the short-circuit current density (Jsc) and fill factor (FF). Enhanced Jsc of 22.25 mA/cm2 and FF of 61.48% were obtained for OPVs based on PTBFTT-Cl, compared to those of PTBFTT-F yielding Jsc of 19.15 mA/cm2 and FF of 57.19%. The external quantum efficiency (EQE) spectra of the corresponding OPVs are illustrated in Fig. 2b, revealing PTBFTT-Cl-based OPVs have broadened EQE response and higher EQE intensities, over that of PTBFTT-F devices. The calculated Jsc values from the integral of EQE spectra for PTBFTT-F and PTBFTT-Cl-based OPVs are 18.61 and 21.51 mA/cm2, which agree with the measured values within the error range of 5%. PTBFTT-Cl-based devices exhibit generally higher EQE response than those of PTBFTT-F-based devices in the broad range of 300–950 nm, which indicates a more efficient photo-charge generation and collection in PTBFTT-Cl-based devices.
 DownLoad:
CSV
DownLoad:
CSV

|
The relationship of the photocurrent density versus the effective voltage of devices is shown in Fig. 2c, revealing the ratios of Jsc/Jsat are 94.7% and 99.3% for PTBFTT-F and PTBFTT-Cl-based devices, respectively (Jsc is the short-circuit current density and Jsat is the saturation photocurrent density when Veff is equal to 2.5 V).
It indicated that PTBFTT-Cl-based devices possessed more efficient exciton dissociation probabilities over those of PTBFTT-F-based OPVs. The dependence of Voc and Jsc values on light intensity (Plight) was also measured (Fig. 2d and Fig. S5 in Supporting information), wherein the dependence of Voc on Plight is subjected to a slope (n) of kT/q (k is the Boltzmann constant, T is the temperature, and q is the elementary charge) [58]. As shown in Fig. 2d, the slopes of 1.137 and 1.103 were obtained for the PTBFTT-F and PTBFTT-Cl-based devices. By fitting the curve with the power-law equation Jsc ∝ (Plight)α (the closer the α value is to 1, the less biomolecular recombination in the corresponding OPVs), we found that the corresponding value of α are 0.987 and 0.992 for PTBFTT-F and PTBFTT-Cl-based devices, respectively. Charge transport properties were examined using hole- and electron-only devices according to the space charge limited current (SCLC) technique (Fig. S5). The measured mobilities of the hole (μh) and electron (μe) were 8.76 × 10−5/4.53 × 10−4 and 9.78 × 10−5/7.66 × 10−4 cm2 V−1 s−1 for PTBFTT-F and PTBFTT-Cl-based blends, respectively. Therefore, PTBFTT-Cl-based OPVs exhibit the improved exciton dissociation probabilities and charge mobilities that are corresponding to the attainable high Jsc and EQE values.
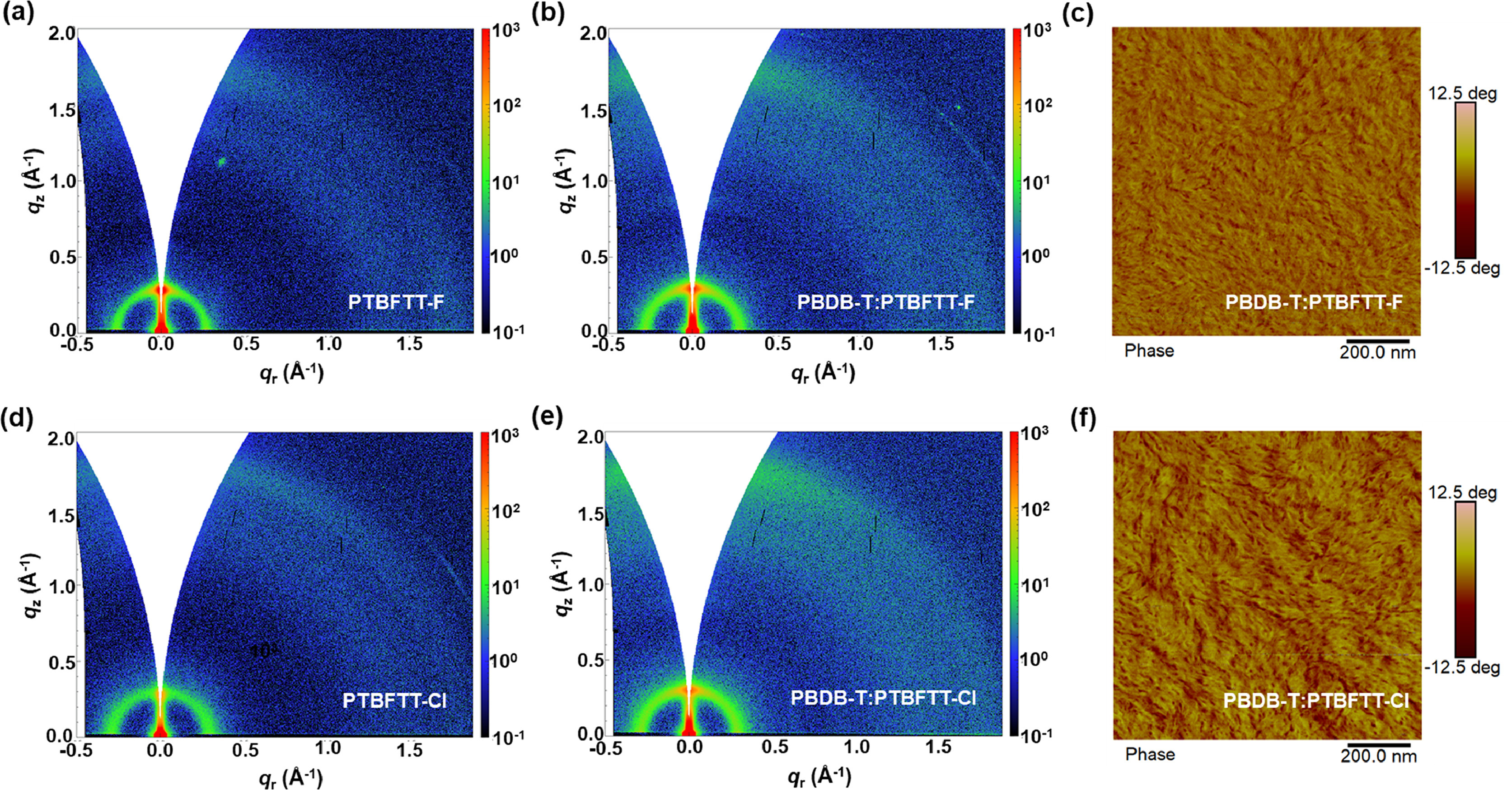
Two-dimensional grazing-incidence wide-angle X-ray scattering (2D-GIWAXS) measurements were employed to investigate the molecular stacking and orientation in neat films and blends (Fig. 3, Figs. S6 and S7, Table S2 in Supporting information). PTBFTT-Cl films show the preferable face-on orientation and densified stacking than those of PTBFTT-F. As for the neat PTBFTT-Cl films, the π-π stacking peak is at 1.79 Å−1 (d = 3.51 Å) in out-of-plane (OOP) direction and lamellar peak is at 0.295 Å−1 (d = 21.3 Å) in-plane (IP) direction. Whereas π-π stacking peak is at 1.72 Å−1 (d = 3.65 Å) in OOP direction and lamellar peak is at 0.270 Å−1 (d = 23.3 Å) in IP direction for the neat PTBFTT-F films. Similarly, as shown in Fig. S7 and Table S2, the PTBFTT-Cl blend films show densified stacking than PTBFTT-F based blends. The surface morphologies of blend films were measured by the atomic force microscopy (AFM) (Fig. 3 and Fig. S8 in Supporting information). The top surface of PTBFTT-F and PTBFTT-Cl blend films exhibit the root-mean-square (RMS) roughness of 0.98 and 2.74 nm, respectively. The relatively rough surface of PTBFTT-Cl blend films would be due to the tendency of strong stacking of PTBFTT-Cl acceptors.
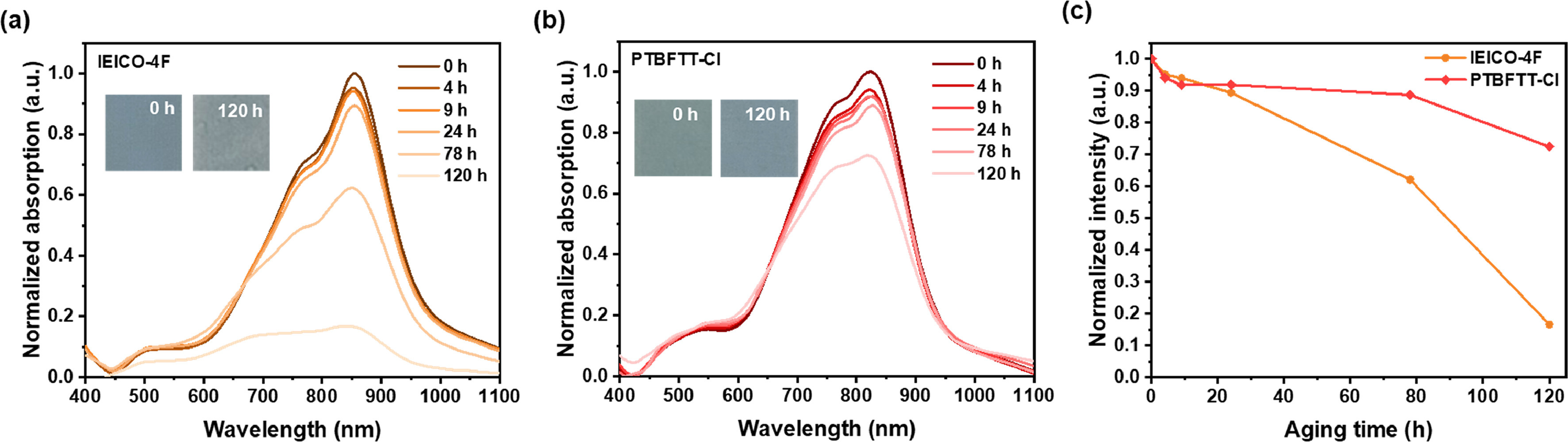
Considering the improved photovoltaic properties, we further investigated the intrinsic photochemical stability of PTBFTT-Cl films under one sun equivalent illumination in ambient. Classic fused NIR acceptor, IEICO-4F [19], was chosen as reference for comparison. It revealed that the non-fused PTBFTT-Cl possessed the superior photochemical stability over that of fused IEICO-4F. The absorption profiles of these samples have been recorded, as shown in Fig. 4. The photodegradation rates of neat films were calculated to be 6.92 × 10−3 h−1 (IEICO-4F) and 2.33 × 10−3 h−1 (PTBFTT-Cl) during the first 120 h illumination in ambient. The dramatic decay of maximum absorption (Amaxabs) is observed for IEICO-4F, whereas PTBFTT-Cl films remain 72% of original absorption intensities for the continuous 120 h illumination. The good photostability of PTBFTT-Cl may stem from the structural factors of NFRA and the capability of dense stacking in solid films.
In conclusion, we have developed two new NIR-NFRAs, PTBFTT-F and PTBFTT-Cl with the design of A-πA-πD-D-πD-πA-A non-fused ring structures. It is found that the introducing electron deficient π-bridge (πA) FTT and multi-noncovalent interactions effectively mediate the planarity and rigidity of NFRAs, extending strong NIR absorption up to 950 nm. The chlorinated acceptors possessing the enlarged π-surface area over that of fluorinated ones, facilitate the molecular stacking and ordering in solid films, which are beneficial to improve the exciton and charge dynamics of corresponding OPVs. OPVs based on PTBFTT-Cl obtained the 10.26% PCE, outperforming those of PTBFTT-F-based devices. In addition, PTBFTT-Cl exhibits the desirable photochemical stability under one sun equivalent illumination in ambient. Overall, this work provides new insights on the design of NIR non-fused electron acceptors.
This research was funded by National Natural Science Foundation of China (No. 22125901), the National Key Research and Development Program of China (No. 2019YFA0705900), the Fundamental Research Funds for the Central Universities (No. 226–2023–00113).
Supplementary material associated with this article can be found, in the online version, at doi:
A. Armin, W. Li, O.J. Sandberg, et al., Adv. Energy Mater. 11 (2021) 2003570. doi: 10.1002/aenm.202003570
Y. Liu, B. Liu, C.Q. Ma, et al., Sci. China Chem. 65 (2022) 224–268. doi: 10.1007/s11426-021-1180-6
S. Karuthedath, J. Gorenflot, Y. Firdaus, et al., Nat. Mater. 20 (2021) 378–384. doi: 10.1038/s41563-020-00835-x
Q. Shen, C. He, S. Li, et al., Acc. Mater. Res. 3 (2022) 644–657. doi: 10.1021/accountsmr.2c00052
Q.Q. Zhang, C.Z. Li, Chem. Eng. J. 452 (2023) 139312. doi: 10.1016/j.cej.2022.139312
J. Yuan, Y. Zhang, L. Zhou, et al., Joule 3 (2019) 1140–1151. doi: 10.1016/j.joule.2019.01.004
L. Zhu, M. Zhang, J. Xu, et al., Nat. Mater. 21 (2022) 656–663. doi: 10.1038/s41563-022-01244-y
J. Gao, N. Yu, Z. Chen, et al., Adv. Sci. 9 (2022) 2203606. doi: 10.1002/advs.202203606
L. Zhan, S. Li, Y. Li, et al., Adv. Energy Mater. 12 (2022) 2201076. doi: 10.1002/aenm.202201076
T. Chen, S. Li, Y. Li, et al., Adv. Mater. 35 (2023) 2300400. doi: 10.1002/adma.202300400
X. Xu, W. Jing, H. Meng, et al., Adv. Mater. 35 (2023) 2208997. doi: 10.1002/adma.202208997
C.J. Traverse, R. Pandey, M.C. Barr, R.R. Lunt, Nat. Energy 2 (2017) 849–860. doi: 10.1038/s41560-017-0016-9
D. Wang, H. Liu, Y. Li, et al., Joule 5 (2021) 945–957. doi: 10.1016/j.joule.2021.02.010
D. Wang, Y. Li, G. Zhou, et al., Energy Environ. Sci. 15 (2022) 2629–2637. doi: 10.1039/D2EE00977C
J. Ren, X. Shu, Y. Wang, et al., Chin. Chem. Lett. 33 (2022) 1650–1658. doi: 10.1016/j.cclet.2021.10.052
X. Li, R. Xia, K. Yan, et al., ACS Energy Lett. 5 (2020) 3115–3123. doi: 10.1021/acsenergylett.0c01554
Y. Li, C. He, L. Zuo, et al., Adv. Energy Mater. 11 (2021) 2003408. doi: 10.1002/aenm.202003408
H. Yao, Y. Chen, Y. Qin, et al., Adv. Mater. 28 (2016) 8283–8287. doi: 10.1002/adma.201602642
H. Yao, Y. Cui, R. Yu, et al., Angew. Chem. Int. Ed. 56 (2017) 3045–3049. doi: 10.1002/anie.201610944
F.X. Chen, J.Q. Xu, Z.X. Liu, et al., Adv. Mater. 30 (2018) 1803769. doi: 10.1002/adma.201803769
Z. Xiao, X. Jia, D. Li, et al., Sci. Bull. 62 (2017) 1494–1496. doi: 10.1016/j.scib.2017.10.017
Z. Jia, S. Qin, L. Meng, et al., Nat. Commun. 12 (2021) 178. doi: 10.1038/s41467-020-20431-6
W. Liu, S. Sun, S. Xu, et al., Adv. Mater. 34 (2022) 2200337. doi: 10.1002/adma.202200337
J. Li, H. Li, L. Ma, S. Zhang, J. Hou, Chin. J. Chem. 41 (2023) 424–430. doi: 10.1002/cjoc.202200579
H. Li, J. Li, C. Yang, et al., J. Mater. Chem. C 11 (2023) 6155–6161. doi: 10.1039/D3TC00625E
Z. Jia, Q. Ma, Z. Chen, et al., Nat. Commun. 14 (2023) 1236. doi: 10.1038/s41467-023-36917-y
T. Li, S. Dai, Z. Ke, et al., Adv. Mater. 30 (2018) 1705969. doi: 10.1002/adma.201705969
Z. Yao, X. Liao, K. Gao, et al., J. Am. Chem. Soc. 140 (2018) 2054–2057. doi: 10.1021/jacs.7b13239
Y. Li, X. Liu, F.P. Wu, et al., J. Mater. Chem. A 4 (2016) 5890–5897. doi: 10.1039/C6TA00612D
S. Liu, J. Yuan, W. Deng, et al., Nat. Photonics 14 (2020) 300–305.
C. He, Y. Li, Y. Liu, et al., J. Mater. Chem. A 8 (2020) 18154–18161. doi: 10.1039/D0TA06907H
Y. Zhou, M. Li, H. Lu, et al., Adv. Funct. Mater. 31 (2021) 2101742. doi: 10.1002/adfm.202101742
X. Wang, R. Zeng, H. Lu, et al., Chin. J. Chem. 41 (2023) 665–671. doi: 10.1002/cjoc.202200673
Y. Liu, Z. Zhang, S. Feng, et al., J. Am. Chem. Soc. 139 (2017) 3356–3359. doi: 10.1021/jacs.7b00566
S. Li, L. Zhan, F. Liu, et al., Adv. Mater. 30 (2018) 1705208. doi: 10.1002/adma.201705208
Z. Zhang, S. Zhang, Z. Liu, et al., Acta Phys. Chim. Sin. 35 (2019) 394–400. doi: 10.3866/PKU.WHXB201805091
Z.P. Yu, Z.X. Liu, F.X. Chen, et al., Nat. Commun. 10 (2019) 2152. doi: 10.1038/s41467-019-10098-z
H. Huang, Q. Guo, S. Feng, et al., Nat. Commun. 10 (2019) 3038. doi: 10.1038/s41467-019-11001-6
T.J. Wen, Z.X. Liu, Z. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 12964–12970. doi: 10.1002/anie.202101867
L. Ma, S. Zhang, J. Zhu, et al., Nat. Commun. 12 (2021) 5093. doi: 10.1038/s41467-021-25394-w
M. Yang, W. Wei, X. Zhou, Z. Wang, C. Duan, Energy Mater. 1 (2021) 100008.
S.Y. Liu, D. Wang, T.J. Wen, et al., Chin. J. Polym. Sci. 40 (2022) 944–950. doi: 10.1007/s10118-022-2750-0
T.J. Wen, J. Xiang, N. Jain, et al., J. Energy Chem. 70 (2022) 576–582. doi: 10.1016/j.jechem.2022.03.030
R. Hou, M. Li, X. Ma, et al., ACS Appl. Mater. Interfaces 12 (2020) 46220–46230. doi: 10.1021/acsami.0c13993
X. Zhang, L. Qin, J. Yu, et al., Angew. Chem. Int. Ed. 60 (2021) 12475–12481. doi: 10.1002/anie.202100390
Q. Shen, C. He, B. Wu, et al., Chem. Eng. J. 471 (2023) 144472. doi: 10.1016/j.cej.2023.144472
D.L. Ma, Q.Q. Zhang, C.Z. Li, Angew. Chem. Int. Ed. 62 (2023) e202214931. doi: 10.1002/anie.202214931
L. Ma, S. Zhang, J. Ren, et al., Angew. Chem. Int. Ed. 62 (2022) e202214088.
Y. Shao, R. Sun, W. Wang, et al., Sci. China Chem. 66 (2023) 1101–1110. doi: 10.1007/s11426-022-1502-3
C. Li, X. Zhang, N. Yu, et al., Adv. Funct. Mater. 32 (2021) 2108861.
C. He, Y. Li, S. Li, et al., ACS Appl. Mater. Interfaces 12 (2020) 16700–16706. doi: 10.1021/acsami.0c00837
T.J. Wen, D. Wang, L. Tao, et al., ACS Appl. Mater. Interfaces 12 (2020) 39515–39523. doi: 10.1021/acsami.0c12100
Q. Wen, Q. Cai, P. Fu, et al., Chin. Chem. Lett. 34 (2023) 107592. doi: 10.1016/j.cclet.2022.06.015
Y. Sakamoto, S. Komatsu, T. Suzuki, J. Am. Chem. Soc. 123 (2001) 4643–4644. doi: 10.1021/ja015712j
S. Feng, M. Li, N. Tang, et al., ACS Appl. Mater. Interfaces 12 (2020) 4638–4648. doi: 10.1021/acsami.9b18076
Z.X. Liu, Z.P. Yu, Z. Shen, et al., Nat. Commun. 12 (2021) 3049. doi: 10.1038/s41467-021-23389-1
X. Zhang, C. Li, L. Qin, et al., Angew. Chem. Int. Ed. 60 (2021) 17720–17725. doi: 10.1002/anie.202106753
X. Zhang, G. Li, S. Mukherjee, et al., Adv. Energy Mater. 12 (2021) 2102172.

Figure 1 (a) Chemical structure with the indicated non-covalent interactions. (b) Isosurfaces of the electrostatic potential (ESP), (c) UV–vis absorption spectra, (d) cyclic voltammetry diagram and (e) thermogravimetric analysis of molecules.

Figure 2 (a) J-V curves and (b) EQE spectra, (c) Jph versus Veff, (d) Voc versus light intensity characteristics for the corresponding OPVs associated with NIR-NFRAs.

Figure 3 2D GIWAXS patterns for (a) PTBFTT-F, (d) PTBFTT-Cl, (b) PBDB-T: PTBFTT-F and (e) PBDB-T: PTBFTT-Cl films. AFM phase images of (c) PBDB-T: PTBFTT-F and (f) PBDB-T: PTBFTT-Cl.

Figure 4 The change of UV–vis absorption spectra under one-sun equivalent illumination in ambient for (a) IEICO-4F and (b) PTBFTT-Cl. (c) The change of absorption intensity versus photoaging time.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载:
 下载:
下载: