
Scheme 1.
azo-DA Reaction, annulations of indoles and 2-vinylindoles, and this work.
Catalytic asymmetric dearomative azo-Diels–Alder reaction of 2-vinlyindoles
Yu-Hang Miao , Zheng-Xu Zhang , Xu-Yi Huang , Yuan-Zhao Hua , Shi-Kun Jia , Xiao Xiao , Min-Can Wang , Li-Ping Xu , Guang-Jian Mei

Rapid preparation of complex cyclic structures, which are wildly found in drug molecules and natural products, from readily accessible starting materials in a catalytic asymmetric manner is one of the central themes in synthetic chemistry. The catalytic asymmetric hetero-Diels–Alder reaction represents among the powerful methodologies in the chemistry toolbox for enantioselective synthesis of six-membered heterocycles [1-4]. As an important variant, azo-Diels–Alder (azo-DA) reaction employs diazenes as dienophiles to construct biologically interesting piperazines (Scheme 1a) [5]. Due to the high electrophilic nature of azo-dienophiles, azo-DA reaction proceeds rapidly even without the need of a catalyst and is therefore regarded as the "click reaction" [6-9]. But conversely, this spontaneity causes strong background reaction and poses a daunting challenge to chemists for developing the catalytic asymmetric version [10]. So far, only sporadic successful examples have been reported by Yamamoto [11], Gong [12-14] and Terada [15]. In their pioneering reports, low temperature strategy (−40 ℃ to −78 ℃) was employed to ensure the high enantioselectivity. Despite these achievements, catalytic asymmetric azo-DA reaction is still in its infancy. It is of great significance for organic synthesis to develop new catalytic asymmetric azo-DA reactions, especially those at ambient temperature.
The catalytic asymmetric dearomatization (CADA, coined by You) reactions have offered a practical strategy for direct assembly of high-value-added three-dimensional polycyclic molecules from readily available planar aromatic feedstocks in an enantio-enriched form [16-19]. In particular, CADA annulation reactions of indole derivatives have developed rapidly since it allows easy access to synthetically and biologically important indolenines and indolines [20-23]. The typical reaction mode involves the disruption of indole aromaticity by making use of the C2=C3 bond as 2π unit. As such, many elegant asymmetric CADA annulation reactions of indoles have been established, which include (2 + 2), (2 + 3), as well as (2 + 4) reactions (Scheme 1b) [24-31]. As an important sub-branch of indole derivatives, 2-vinylindoles recently have been utilized as versatile platform molecules in asymmetric synthesis [32-35]. Bearing a C3-substituent, 2-vinylindoles usually behave as mono-olefins by switching the nucleophilic site from the C3- to the C2"-position (Scheme 1b). For instance, Shi and Schneider reported the chiral phosphoric acid (CPA) catalyzed (2 + n) reactions of 3-alkyl-2-vinylindoles to constructing enantioenriched indole-containing compounds [36-42]. However, when it comes to dearomatization, 2-vinylindoles have been utilized as 2,3-disubstituted indoles and hardly undergo the vinylogous CADA (4 + n) reaction [43], [44]. To our knowledge, the only example comes from the MacMillan group, in which they achieved the collective total synthesis of indole alkaloids via organocatalytic asymmetric (4 + 2) cascade reactions of tryptamine-derived 2-vinylindoles [45-48]. Consequently, although challenging, developing dearomative asymmetric (4 + n) reactions of 2-vinylindoles will open up new opportunities for synthetic chemistry.
To address the above issues, we envision that merging catalytic asymmetric dearomatization of 2-vinylindoles and azo-DA reaction is feasible, wherein the required high energy barrier for dearomatization could avert the strong background reaction of azo-DA reaction (Scheme 1c). Our group has a long-term interest in developing facile methods for the synthesis of azo-compounds [49-53]. Along this line, we report herein the CPA-catalyzed asymmetric dearomative azo-DA reaction between 2-vinylindoles 1 as dienes and common triazolediones 2 as dienophiles under mild conditions, which delivers a facile access to a variety of piperazine-fused tetracyclic indole derivatives 3 in good yields and with excellent stereoselectivities. Density functional theory (DFT) calculations have been performed to gain insights into the reaction mechanism and the origins of the enantioselectivity. This protocol not only represents the first catalytic asymmetric dearomative azo-Diels–Alder reaction performed at room temperature, but also enriches the reaction of 2-vinylindoles.
Our investigation was initiated by employing the dearomative azo-DA reaction of 2-vinylindole 1a and triazoledione 2a as model reaction (Table 1). To our delight, the projected reaction occurred smoothly at room temperature under the catalysis of CPA-1, delivering the dearomative [4 + 2] cycloaddition product 3a in good yield with excellent diastereoselectivity (entry 1). Then, solvent effect was studied (entries 1−4). Among them, tetrahydrofuran (THF) gave an encouraging result for enantio-control (entry 3). So, THF was selected as the best solvent for the next catalyst screening [54-56]. While TRIP-CPA was less effective (entry 5), CPA-3 and −4 led to slightly superior results (entries 6 and 7). Further examinations demonstrated that binol-derived CPA-5 and −6 failed to increase the ee value (entries 8 and 9). Finally, by using the spinol-derived CPA-7 as catalyst, this asymmetric dearomative azo-DA reaction took place in an excellent yield and with excellent diastereo- and enantio-selectivity (97% yield, > 20:1 dr, 92% ee, entry 10). It is worth noting that due to the observed background reaction, it was necessary to add 2a in batches to ensure the excellent yield and enantioselectivity (entry 11), (S)-CPA 1–6 and (S)-CPA-7 gave the enantiomers of 3a.
With the optimal conditions in hand, the substrate scope was then investigated. As shown in Scheme 2, this CPA-catalyzed dearomatization reaction was applicable to a variety of 2-vinylindoles 1. It was found that substituents on the phenyl ring of indole motif (R) were all compatible, furnishing products 3b−3g in consistently excellent ee values. The position of the substituent had some delicate influence on the yield. For instance, among the bromo-substituted cases, C7-bromo-substrate afforded a lower yield than C5- and C6-subustituted ones (3g vs. 3c & 3f). In addition, the variation of the alkyl group at the C3-position of indole motif was tested, which was significant to the construction of aza-quaternary carbon centers. Ethyl and benzyl groups were tolerated, giving products 3h−3i in good yields and with high levels of enantioselectivity. Besides, 2-vinylindoles bearing various R2 groups, such as substituted phenyl and naphthyl, served as suitable substrates, and the corresponding products 3j−3r were obtained in good to excellent yields and with nearly perfect ee values. More importantly, changing the aryl group appended on the double bond to the alkyl group could be conducted with the retention of excellent enantioselectivity (3s). Notably, in all cases tested, excellent diastereoselectivities were observed (all dr > 20:1).

Next, the substrate scope of triazolediones 2 was investigated (Scheme 3). Various N-substituted triazolediones had been engaged in the reaction. For N-aryl triazolediones, the position and electronic nature of the substituents on the phenyl ring had little effect on the reaction. All the tetracyclic products 3t−3e′ were formed in excellent yields with excellent enantioselectivities (90%–97% yields, 90%–98% ee). Notably, the absolute configuration of the product 3z was assigned based on X-ray crystallographic analysis. Interestingly, products 3z−3e′ potentially process a C−N axial chirality, indicating that this reaction could be applied to the desymmetrization of triazolediones [57]. However, density functional theory (DFT) calculations of 3z showed an unstable conformation with a rotational barrier of 20.1 kcal/mol. Triazoledione with a bulky ortho-tert-butyl group was then employed. Under further optimized conditions (see Supporting information for details), 3f′ with a rotationally stable C−N axis was created in a good yield and with a high stereoselectivity. Furthermore, N-benzyl group was also well tolerated, delivering the product 3g′ in consistently good results.

To gain insights into the reaction mechanism, control experiments were performed (Scheme 4). Firstly, changing azo-dienophile triazoledione to common dienophile pyrroledione 4 was not feasible (Scheme 4a) [58]. The reasons could be attributed to the higher energy barrier of alkylation than amination and the lack of hydrogen-bonding site. When N-methyl-2-vinylindole 5 was utilized, instead of the desired cycloaddition product, a complex result was observed, which further indicated the importance of hydrogen-bonding network in the transition state (Scheme 4b). The nonlinear effect experiment was then carried out (Scheme 4c). The perfect linear relationship between catalyst and product implied that the enantio-determining step involved only one component of CPA. When acyclic azodicarboxylates 6 was utilized, only the dearomative hydrazination (7′) at the C3-position was observed (Scheme 4d) [59]. Under standard conditions, the hydrazination product 7′ cannot be transformed into the cycloaddition product 7. Other available azodicarboxylates were also examined, and similar unstable C3-hydrazination products were obtained. On the basis of these experimental results, the dual-hydrogen-bonding network was suggested, which led to two plausible reaction pathways a and b (Scheme 4e). While pathway a showed the C3-nucleophilicity of indoles, pathway b involved the C2"-nucleophilicity.

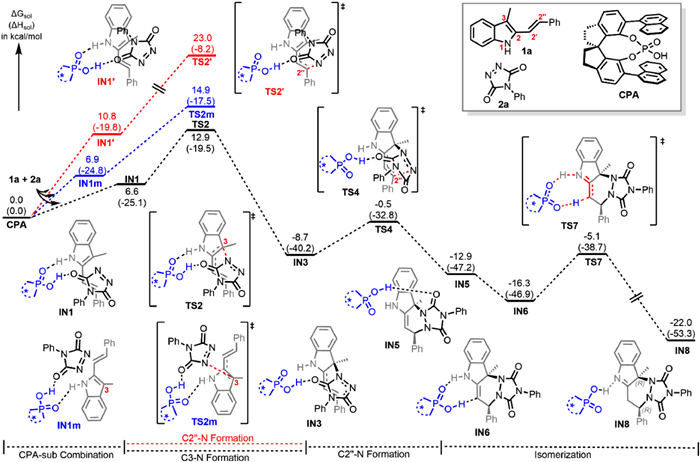
Additionally, DFT calculations were conducted with the B3LYP-D3(BJ) functional to shed light on the reaction mechanism and the origins of the enantioselectivity in the CPA-catalyzed asymmetric dearomatization reaction of 2-vinylindoles (see Supporting information for computational details). First of all, the concerted vs. stepwise, and endo vs exo Diels-Alder reaction pathways were all calculated, and the stepwise, endo pathway was found to be most favorable (see Supporting information for more details). Two possibilities for the stepwise endo pathway were considered: the C3-N bond formation occurring firstly or the C2"-N bond formation occurring firstly. The calculated free energy profile is shown in Fig. 1. The binding of 1a and 2a to CPA through hydrogen bonding leads to IN1. The C3-N formation occurs via TS2 to generate intermediate IN3, which bears a free energy barrier of 12.9 kcal/mol with respect to CPA + 1a +2a. Subsequently, the C2"-N formation aided by the CPA catalyst takes place to generate the cyclic intermediate IN5, requiring a free energy barrier of 8.2 kcal/mol with respect to IN3. Rearrangement of the CPA motif leads to intermediate IN6, followed by CPA-assisted enamine-to-imine tautomerization via TS7 to generate the RR-chirality product IN8, which demands 11.2 kcal/mol free energy. On the other hand, for the pathway with the C2"-N bond formation occurring firstly (red path in Fig. 1), it has the activation free energy barrier of 23.0 kcal/mol (via TS2′), which is 10.1 kcal/mol unfavorable than that with C3-N formation occurring firstly (12.9 kcal/mol via TS2). Thus, the most favorable pathway for the CPA-catalyzed Diels Alder reaction involves the complexation, C3-N formation, C2"-N formation, and tautomerization steps, and among which the C3-N formation is the rate-determining step. To better understand the origins of enantioselectivity, we also calculated the rate-determining intermediate and transition state that led to the formation of the minor enantiomer (blue path in Fig. 1). It shows that the C3-N formation occurring via TS2m has a free energy barrier of 14.9 kcal/mol, which is 2.0 kcal/mol higher compared to the black pathway that leads to the major RR-enantiomer (via TS2, 12.9 kcal/mol). Thus, the calculated ee (93%) agrees well with the experimental value (92%).
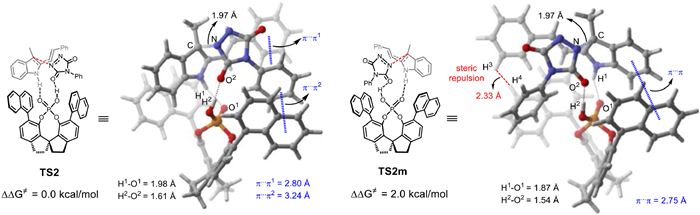
By detailed analysis of the selectivity-determining transition states, it is found that in TS2m, there is steric repulsion between substrate 1a and the CPA catalyst, as manifested by the close H3-H4 distance (2.33 Å, Fig. 2). In addition, in TS2, the phenyl rings of 1a, 1b, and the naphthalene ring of the CPA forms a sandwich-like structure in which two favorable π…π interactions are formed (i.e., π…π1: 2.80 Å; π…π2: 3.24 Å). By contrast, there is only one π…π interaction found in TS2m (π…π: 2.75 Å). This is also supported by the independent gradient model based on Hirshfeld partition (IGMH) analysis [60,61] for TS2 and TS2m (see Supporting information for details), in which the stronger π…π interactions are observed in the former than the latter. Furthermore, distortion-interaction analysis (see Supporting information for details) [62-65] shows that the relative distortion energy in TS2m is 2.0 kcal/mol larger than TS2, and the interaction energy in TS2 is 0.8 kcal/mol stronger than TS2m, which supports the above-mentioned steric and non-covalent interaction arguments. Thus, the large steric repulsion and the relatively less π…π interaction that disfavoring TS2m account for the origins of the high enantioselectivity.
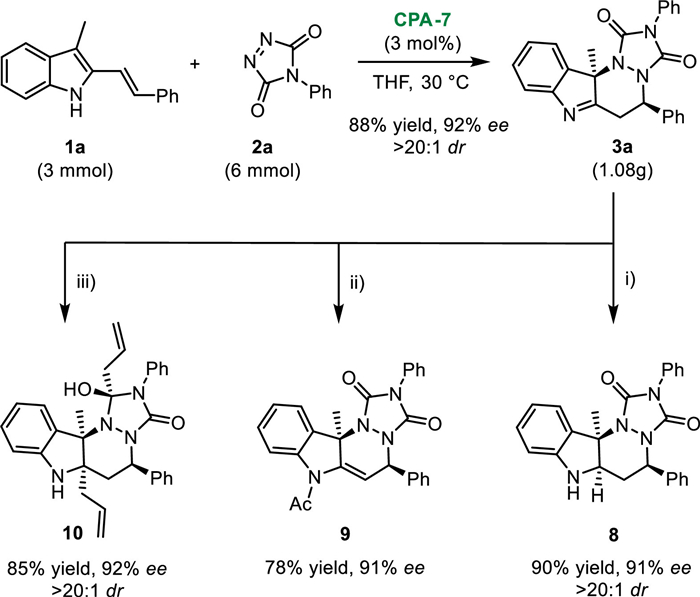
Finally, to demonstrate the synthetic utility of this protocol, some preliminary applications were performed (Scheme 5). Firstly, this dearomative azo-DA reaction could be conducted on 3.0 mmol scale under standard conditions, affording the desired product 3a in 88% yield (1.08 g) with 92% ee and > 20:1 dr. Besides, the reduction of imine motif with NaBH4 led to the formation of 8 in a good yield, albeit with a slight erosion of enantiomeric purity. N-protection with acetyl group was performed to give the double migration product 9. Interestingly, both the imine and carbonyl groups could go through 1,2-addition reaction with allyl magnesium bromide, delivering the product 10 in 85% yield with 92% ee and > 20:1 dr.
In conclusion, we have established the first catalytic asymmetric dearomative azo-Diels–Alder reaction of 2-vinylindoles with triazolediones. Merging catalytic asymmetric dearomatization and azo-Diels–Alder reaction allows the implementation of the projected reaction at mild conditions, since the required high energy barrier for dearomatization could prevent the strong background reaction of azo-Diels–Alder reaction. Density functional theory calculations were conducted to shed light on the reaction mechanism and the origins of the enantioselectivity. By using this method, a variety of tetracyclic indole derivatives were readily prepared in good to excellent yields and with excellent diastereo- and enantio-selectivity. The evaluation of the biological activities of these synthesized products is currently ongoing in our laboratory and will be reported in due course.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful for the generous financial support from Natural Science Foundation of Henan Province (No. 222300420084), application research plan of Key Scientific Research Projects in Colleges and Universities of Henan Province (No. 22A150056), the Youth Innovation Team Program in Colleges and Universities of Shandong Province (No. 2022KJ228), and National Natural Science Foundation of China (No. 22208302).
Supplementary material associated with this article can be found, in the online version, at doi:
K.A. Jørgensen, Angew. Chem. Int. Ed. 39 (2000) 3558–3588. doi: 10.1002/1521-3773(20001016)39:20<3558::AID-ANIE3558>3.0.CO;2-I
L. Lin, X. Liu, X. Feng, Synlett 2007 (2007) 2147–2157. doi: 10.1055/s-2007-984917
V. Eschenbrenner-Lux, K. Kumar, H. Waldmann, Angew. Chem. Int. Ed. 53 (2014) 11146–11157. doi: 10.1002/anie.201404094
M.G. Vinogradov, O.V. Turova, S.G. Zlotin, Adv. Synth. Catal. 363 (2021) 1466–1526. doi: 10.1002/adsc.202001307
U. Mäeorg, S. Tšupova, Heterocycles 88 (2014) 129. doi: 10.3987/REV-13-SR(S)3
S. Billiet, K. De Bruycker, F. Driessen, et al., Nat. Chem. 6 (2014) 815–821. doi: 10.1038/nchem.2023
L. Crouillebois, L. Pantaine, J. Marrot, et al., J. Org. Chem. 80 (2015) 595–601. doi: 10.1021/jo502087a
L. Xiao, Y. Chen, K. Zhang, Macromolecules 49 (2016) 4452–4461. doi: 10.1021/acs.macromol.6b00782
T. Varlet, G. Levitre, P. Retailleau, G. Masson, Bioorg. Med. Chem. 27 (2019) 2438–2443. doi: 10.1016/j.bmc.2019.02.008
P.S. Aburel, W. Zhuang, R.G. Hazell, K.A. Jorgensen, Org. Biomol. Chem. 3 (2005) 2344–2349. doi: 10.1039/b503744a
M. Kawasaki, H. Yamamoto, J. Am. Chem. Soc. 128 (2006) 16482–16483. doi: 10.1021/ja066726y
B. Liu, K.N. Li, S.W. Luo, et al., J. Am. Chem. Soc. 135 (2013) 3323–3326. doi: 10.1021/ja3110472
B. Liu, T.Y. Liu, S.W. Luo, L.Z. Gong, Org. Lett. 16 (2014) 6164–6167. doi: 10.1021/ol503047s
Y.L. Du, F. Zhao, Z.Y. Han, L.Z. Gong, Synthesis 49 (2017) 151–158.
N. Momiyama, H. Tabuse, H. Noda, et al., J. Am. Chem. Soc. 138 (2016) 11353–11359. doi: 10.1021/jacs.6b07150
C.X. Zhuo, W. Zhang, S.L. You, Angew. Chem. Int. Ed. 51 (2012) 12662–12686. doi: 10.1002/anie.201204822
W.T. Wu, L. Zhang, S.L. You, Chem. Soc. Rev. 45 (2016) 1570–1580. doi: 10.1039/C5CS00356C
Y.Z. Cheng, X. Zhang, S.L. You, Sci. Bull. 63 (2018) 809–811. doi: 10.1016/j.scib.2018.06.006
C. Zheng, S.L. You, ACS Cent. Sci. 7 (2021) 432–444. doi: 10.1021/acscentsci.0c01651
C. Zheng, S.L. You, Nat. Prod. Rep. 36 (2019) 1589–1605. doi: 10.1039/C8NP00098K
F.T. Sheng, J.Y. Wang, W. Tan, Y.C. Zhang, F. Shi, Org. Chem. Front. 7 (2020) 3967–3998. doi: 10.1039/D0QO01124J
G.J. Mei, W.L. Koay, C.X.A. Tan, Y. Lu, Chem. Soc. Rev. 50 (2021) 5985–6012. doi: 10.1039/D0CS00530D
Y.Z. Liu, H. Song, C. Zheng, S.L. You, Nat. Synth. 1 (2022) 203–216. doi: 10.1038/s44160-022-00039-y
Z.L. Niemeyer, S. Pindi, D.A. Khrakovsky, et al., J. Am. Chem. Soc. 139 (2017) 12943–12946. doi: 10.1021/jacs.7b08791
L.W. Qi, J.H. Mao, J. Zhang, B. Tan, Nat. Chem. 10 (2018) 58–64. doi: 10.1038/nchem.2866
Q. Yu, J. Huang, J. Qin, H. Zuo, Y. Wu, F. Zhong, ACS Catal. 9 (2019) 7285–7291. doi: 10.1021/acscatal.9b01734
G.J. Mei, X. Tang, Y. Tasdan, Y. Lu, Angew. Chem. Int. Ed. 59 (2020) 648–652. doi: 10.1002/anie.201911686
L. Shen, Y. Zheng, Z. Lin, et al., Angew. Chem. Int. Ed. 62 (2023) e202217051. doi: 10.1002/anie.202217051
M.C. Tong, X. Chen, J. Li, et al., Angew. Chem. Int. Ed. 53 (2014) 4680–4684. doi: 10.1002/anie.201400109
L.W. Feng, H. Ren, H. Xiong, et al., Angew. Chem. Int. Ed. 56 (2017) 3055–3058. doi: 10.1002/anie.201611734
W. Shao, S.L. You, Chem. Eur. J. 23 (2017) 12489–12493. doi: 10.1002/chem.201703443
G.J. Mei, F. Shi, Synlett 27 (2016) 2515–2524. doi: 10.1055/s-0036-1588611
E. Rossi, G. Abbiati, V. Pirovano, Eur. J. Org. Chem. 2017 (2017) 4512–4529. doi: 10.1002/ejoc.201700120
M.S. Tu, K.W. Chen, P. Wu, et al., Org. Chem. Front. 8 (2021) 2643–2672. doi: 10.1039/D0QO01643H
Y.A.A.M. Elshaier, M.T.M. Nemr, M.S. Refaey, W.A.A. Fadaly, A. Barakat, New J. Chem. 46 (2022) 13383–13400. doi: 10.1039/D2NJ00460G
W. Tan, X. Li, Y.X. Gong, M.D. Ge, F. Shi, Chem. Commun. 50 (2014) 15901–15904. doi: 10.1039/C4CC07246D
J.J. Zhao, S.B. Sun, S.H. He, Q. Wu, F. Shi, Angew. Chem. Int. Ed. 54 (2015) 5460–5464. doi: 10.1002/anie.201500215
K. Bera, C. Schneider, Chem. Eur. J. 22 (2016) 7074–7078. doi: 10.1002/chem.201601020
Z.Q. Zhu, L. Yin, Y. Wang, et al., Org. Chem. Front. 4 (2017) 57–68. doi: 10.1039/C6QO00446F
X.L. Jiang, S.F. Wu, J.R. Wang, G.J. Mei, F. Shi, Adv. Synth. Catal. 360 (2018) 4225–4235. doi: 10.1002/adsc.201800829
I. Kallweit, C. Schneider, Org. Lett. 21 (2019) 519–523. doi: 10.1021/acs.orglett.8b03833
R. Sarkar, I. Kallweit, C. Schneider, Org. Lett. 24 (2022) 6433–6437. doi: 10.1021/acs.orglett.2c02548
Y. Wang, M. Sun, L. Yin, F. Shi, Adv. Synth. Catal. 357 (2015) 4031–4040. doi: 10.1002/adsc.201500901
K.W. Chen, D.D. Wang, S.J. Liu, et al., J. Org. Chem. 86 (2021) 10427–10439. doi: 10.1021/acs.joc.1c01105
S.B. Jones, B. Simmons, D.W.C. MacMillan, J. Am. Chem. Soc. 131 (2009) 13606–13607. doi: 10.1021/ja906472m
S.B. Jones, B. Simmons, A. Mastracchio, D.W. MacMillan, Nature 475 (2011) 183–188. doi: 10.1038/nature10232
B.D. Horning, D.W. MacMillan, J. Am. Chem. Soc. 135 (2013) 6442–6445. doi: 10.1021/ja402933s
B.N. Laforteza, M. Pickworth, D.W. Macmillan, Angew. Chem. Int. Ed. 52 (2013) 11269–11272. doi: 10.1002/anie.201305171
G.J. Mei, J.J. Wong, W. Zheng, et al., Chem 7 (2021) 2743–2757. doi: 10.1016/j.chempr.2021.07.013
T.J. Han, Z.X. Zhang, M.C. Wang, L.P. Xu, G.J. Mei, Angew. Chem. Int. Ed. 61 (2022) e202207517. doi: 10.1002/anie.202207517
Y.H. Miao, Y.Z. Hua, H.J. Gao, et al., Chem. Commun. 58 (2022) 7515–7518. doi: 10.1039/D2CC02458F
C.Y. Guan, T.J. Han, S.K. Jia, Y.Z. Hua, G.J. Mei, Green Synth. Catal. 4 (2023) 258–262. doi: 10.1016/j.gresc.2022.05.003
F.Y. Yang, T.J. Han, S.K. Jia, M. Wang, G.J. Mei, Chem. Commun. 59 (2023) 3107–3110. doi: 10.1039/D3CC00160A
X. Li, Q. Song, Chin. Chem. Lett. 29 (2018) 1181–1192. doi: 10.1016/j.cclet.2018.01.045
X. Xiang, Z. He, X. Dong, Chin. J. Org. Chem. 43 (2023) 791–808. doi: 10.6023/cjoc202211043
W. Yang, H. Wang, Z. Pan, Z. Li, W. Deng, Chin. Chem. Lett. 31 (2020) 721–724. doi: 10.1016/j.cclet.2019.09.008
J.W. Zhang, J.H. Xu, D.J. Cheng, et al., Nat. Commun. 7 (2016) 10677. doi: 10.1038/ncomms10677
S. Liu, J. Wang, Y. Ma, et al., Chin. Chem. Lett. 33 (2022) 2041–2043. doi: 10.1016/j.cclet.2021.09.030
X.Y. Qiu, Z.H. Li, J. Zhou, et al., ACS Catal. 12 (2022) 7511–7516. doi: 10.1021/acscatal.2c02164
T. Lu, Q.X. Chen, J. Comput. Chem. 43 (2022) 539. doi: 10.1002/jcc.26812
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
L.P. Xu, Z. Zhuang, S. Qian, J.Q. Yu, D.G. Musaev, ACS Catal. 12 (2022) 4848–4858. doi: 10.1021/acscatal.2c00879
L.P. Xu, J.B. Roque, R. Sarpong, D.G. Musaev, J. Am. Chem. Soc. 142 (2020) 21140–21152. doi: 10.1021/jacs.0c10220
F.M. Bickelhaupt, K.N. Houk, Angew. Chem. Int. Ed. 56 (2017) 10070–10086. doi: 10.1002/anie.201701486
D.H. Ess, K.N. Houk, J. Am. Chem. Soc. 129 (2007) 10646–10647. doi: 10.1021/ja0734086

Scheme 2 Substrate scope of 2-vinylindoles. Reaction conditions: 1 (0.1 mmol), 2a (0.2 mmol), and (S)-CPA-7 (3 mol%) in THF (1 mL) at r.t. for 2.5 h, isolated yields, ee and dr values were determined by chiral HPLC analysis.

Scheme 3 Substrate scope of triazolediones. Reaction conditions: 1 (0.1 mmol), 2 (0.2 mmol), and (S)-CPA-7 (3 mol%) in THF (1 mL) at r.t. for 2.5 h, isolated yields, ee and dr values were determined by chiral HPLC analysis. a The reaction was performed with (S)-CPA-4.

Figure 1 The free energy profile for the CAP-catalyzed Diels-Alder reaction of 2-vinylindole (1a) and triazoledione (2a) (calculations were performed at the SMD(THF)/M06–2X/6–311++G(d, p)//SMD(THF)/B3LYP-D3(BJ)/6–31G(d, p) level of theory).

Figure 2 The comparison and the geometries of the enantioselectivity-determining transition states, TS2 and TS2m.

Scheme 5 Preliminary applications. Reaction conditions: ⅰ) 3a (0.1 mmol), NaBH4 (0.12 mmol), MeOH, r.t.; ⅱ) 3a (0.1 mmol), acetyl chloride (0.15 mmol), NaH (0.15 mmol), toluene, r.t.; ⅲ) 3a (0.1 mmol), allyl magnesium bromide (0.4 mmol), toluene, 0 ℃., r.t. = room temperature.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

 下载:
下载: