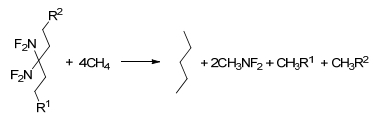
Figure 1.
Molecular frameworks of 3, 3-bis(difluoroamino)- 1, 5-substituted pentane derivatives

The intrinsically high energy of difluoroamino groups has attracted many attentions and interests in organic difluoramines which have been mainly due to their potential as energetic materials in rocket propellants and explosives.[1] Many works on structure design and synthetic method development of difluoroamino-based energetic oxidizers, [2, 3, 4, 5, 6, 7] polymers[8, 9] and plasticizers[10, 11] with stable structures have been done since the late 1980s. Recently, our group have synthesized five difluoroamino energetic polymers with well-designed structure and good thermal stability.[12, 13, 14] To meet the increasing demand for application of these difluoroamino energetic polymers, the design and development of novel difluoroamino plasticizers are worthwhile.
Properties could be often manipulated by making structural modifications. The primary step for design and synthesis of new energetic materials is the optimization of the candidates with high energy and less sensitivity.[15] In the past several decades, theoretical studies based on quantum chemical treatments have been improved as an efficient research tool for structure optimization and property prediction of novel energetic materials.[16~19] They could provide more understandings of the relationships between molecular structure and property without dangerous and expensive experimental tests.
As a promising candidate of difluoroamino-based plasticizers, 3, 3-bis(difluoroamino)-1, 5-dinitratopentane[11] was designed and synthesized with low glass transition temperature and good energy properties. To Figure out if there are other 3, 3-bis(difluoroamino)-1, 5-substituted pentane derivatives with better energy property and low sensitivity, we report a systematic study of the heats of formation (HOFs), energetic properties, and thermal stability of a series of 3, 3-bis(difluoroamino)-1, 5-substituted pentane derivatives with different substituents by using density function theory (DFT).
Figure 1 presents the molecular frameworks of a series of 3, 3-bis(difluoroamino)-1, 5-substituted pentane derivatives. Based on the substituted energetic groups, the compounds can be divided into two series (S symmetric substituted in 1 and 5 position of pentane and A asymmetric substituted in 1 and 5 position of pentane).
Isodesmic reactions could be employed to calculate the heats of formation (HOFs) using total energies obtained from ab initio calculations.[20, 21] Herein, isodesmic reactions (Figure 2) are designed in which the numbers of all types of bonds are invariable. Eq. 1 is used to calculate the heat of reaction ΔH298 at 298 K.
|
$\Delta {H_{298}} = \Delta {H_{{\rm{f,P}}}} - \Delta {H_{{\rm{f,R}}}}$ |
(1) |
Where ΔHf, P and ΔHf, R are the HOFs of products and reactants at 298 K, respectively.
The experimental HOFs of reference compounds CH4, C5H12, CH3ONO2, CH3NO2 and (CH3)2NNO2 are available, and the HOFs of CH3NF2 and CH3N3 are calculated by Ju et al.[22, 23] The value of ΔH298 can be also calculated via Eq. 2.
|
$ \begin{array}{l} \Delta {H_{298}} = \Delta {E_{298}} + \Delta \left( {PV} \right)\\ \;\;\;\;\;\;\;\;\; = \Delta {E_0} + \Delta {E_{{\rm{ZPE}}}} + \Delta {E_{\rm{T}}} + \Delta nRT \end{array} $ |
(2) |
Where ΔE0 is the change in total energy between the products and the reactants at 0 K, ΔEZPE is the difference between the zero-point energies (ZPE) of the products and reactants at 0 K, ΔET is the thermal correction from 0 to 298 K. For the isodesmic reaction in this calculation process, Δn=0, so Δ(PV)=0.
The calculation of detonation properties for most energetic compounds requires solid phase HOF (ΔHf, solid) since their condensed phase is solid. According to Hess's low of constant heat summation[24] and Politzer's research, [25, 26] ΔHf, solid can be calculated via Eqs. 3 and 4.
|
$ \Delta {H_{{\rm{f}},{\rm{solid}}}} = \Delta {H_{{\rm{f}},{\rm{gas}}}} - \Delta {H_{{\rm{sub}}}} $ |
(3) |
|
$ \Delta {H_{{\rm{sub}}}} = a{A^2} + b{(v\sigma _{{\rm{tot}}}^{\rm{2}})^{0.5}} + c $ |
(4) |
Where ΔHf, gas is the gas phase HOF, ΔHsub is heat of sublimation, A is the surface area of the 0.001 electrons/bohr3 isosurface of the electronic density of the molecule, ν is the balance of charges between positive potential and negative potential on the isosurface,
Modified Kamlet-Jacobs equations are used to estimate the detonation velocity and pressure[28, 29] (Eqs. 5 and 6). And the theoretical crystal density is obtained from the Eq. 7 proposed by Polizer et al.[30]
|
$D = 1.01{(N{\overline M ^{1/2}}{Q^{1/2}})^{1/2}}(1 + 1.30\rho )$ |
(5) |
|
$P = 1.558{\rho ^2}N{\overline M ^{1/2}}{Q^{1/2}}$ |
(6) |
|
$\rho = \alpha (\frac{M}{{V(0.001)}}) + \beta v\sigma _{{\rm{tot}}}^{\rm{2}} + \gamma $ |
(7) |
Each term in Eqs. 5~7 is defined as follows: D, detonation velocity (km/s); P, detonation pressure (GPa); N, moles of detonation gases per gram explosives;
The impact sensitivity (h50, cm) is defined as the height from which there is a 50% probability of causing an explosion. Impact sensitivity is generally reported either as this height in cm, designated h50, or as the corresponding impact energy in J. With a 2.5 kg mass, an h50 of 1 cm is equivalent to an impact energy of 0.245 J. The greater the value of h50 or the impact energy, the less is the sensitivity. Eq. 8 presents a simple method to estimate the h50 of energetic compounds.[31]
|
${h_{50}} = \alpha \sigma _{\rm{ + }}^2 + \beta v + \gamma $ |
(8) |
Where
The bond dissociation energy (BDE), which reflects the strength of bonding, is fundamental to understand chemical processes.[32] The BDE and BDE with ZPE correction of reaction A-B(g)→A•(g)+B•(g) at 298 K and 101 kPa are given in terms of Eqs. 9 and 10.[33, 34]
|
$ {\rm{BD}}{{\rm{E}}_0}({\rm{A}} - {\rm{B}}){\rm{ = }}{E_0}\left( {{\rm{A}} \cdot } \right) + {E_0}\left( {{\rm{B}} \cdot } \right) - {E_0}\left( {{\rm{A}} - {\rm{B}}} \right) $ |
(9) |
|
${\rm{BDE}}{({\rm{A}} - {\rm{B}})_{{\rm{ZPE}}}}{\rm{ = BD}}{{\rm{E}}_0}({\rm{A}} - {\rm{B}}) + \Delta {E_{{\rm{ZPE}}}}$ |
(10) |
where ∆EZPE is the difference between the ZPEs of the products and the reactions.
Gaussian 09 package[35] is used to perform the calculations at B3LYP/6-311G++(d, p)[36] and B3PW91/6-31G(d, p)[37] level for different situations. The optimizations are performed without any symmetry restrictions, using the default convergence criteria in the program. All the optimized structures are characterized to the true local energy minima on the potential surface without imaginary frequencies. The total energy, zero-point energy and thermal correction are calculated with vibration frequencies analysis. The volume and surface electrostatic potential are calculated by Multiwfn program.[38]
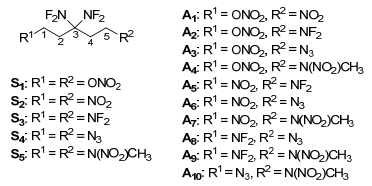
Here, the effects of different substituents in 1 and 5 positions of 3, 3-bis(difluoroamino) pentane on the gas-phase and solid phase HOFs of the title compounds are investigated. Table 1 presents the calculated total energy (E0), zero-point energies (ZPE), thermal corrections (HT) and heat of formation (HOFs) of the reference compounds in the isodesmic reactions at B3LYP/6-311++G(d, p) level. Table 2 shows the calculated E0, ZPE, HT, molecular properties, heat of sublimation and HOFs of the title compounds. S4 has the highest ∆Hf, gas and ∆Hf, solid among the title compounds for the substitution of two azido groups. More clear evidence about the effects of the different substituents on ∆Hf, gas and ∆Hf, solid of the title compounds is presented in Figure 3. The calculated ∆Hf, solid has the same variation trend of ∆Hf, gas. The substitution of N3 enhances the HOF values of the title compounds extremely. HOF values increase with the order of substituent species ONO2 < NF2 < NO2 < N(NO2)CH3≪N3. This can be attributable to the increase of the number of energetic N—N and C—N bonds in the title compounds, which is in agreement with the previous researches[41, 42] that the more N—N bonds a compound has, the higher its HOF is.
 下载:
导出CSV
下载:
导出CSV
| Compd. | E0/a.u. | ZPEa/a.u. | HTa/(kJ•mol-1) | HOF/(kJ•mol-1) |
| H4 | -40.4894 | 0.04365 | 9.61 | -74.60b |
| C5H12 | -197.6702 | 0.15650 | 20.30 | -146.90b |
| CH3NF2 | -294.2638 | 0.04564 | 13.14 | -115.23d |
| CH3ONO2 | -320.2390 | 0.05292 | 15.00 | -124.40b |
| CH3NO2 | -245.0420 | 0.04864 | 13.35 | -74.30b |
| CH3N3 | -204.1033 | 0.04908 | 13.64 | 296.54e |
| (CH3)2NNO2 | -339.6683 | 0.09274 | 19.26 | -0.84c |
| a The scaling factors for ZPE and HT were 0.98 and 0.96, respectively.[43] bExperimental values taken from Ref. [40]. cExperimental values taken from Ref. [39]. d Calculated values taken from Ref. [22]; e Calculated values taken from Ref. [23]. | ||||
下载:
导出CSV
| Compd. | E0/a.u. | ZPEa/a.u. | HT/(kJ•mol-1) | ∆Hf, gas/(kJ•mol-1) | A/A2 | ∆Hsub/(kJ•mol-1) | ∆Hf, solid/(kJ•mol-1) | |
| S1 | -1264.7195 | 0.1697 | 50.21 | -343.57 | 258.68 | 21.85 | 119.43 | -463.00 |
| S2 | -1114.3217 | 0.1617 | 45.92 | -232.92 | 237.41 | 39.48 | 118.75 | -351.67 |
| S3 | -1212.7655 | 0.1555 | 46.70 | -314.63 | 233.80 | 14.86 | 100.09 | -414.72 |
| S4 | -1032.4513 | 0.1625 | 47.05 | 490.73 | 253.08 | 17.10 | 112.51 | 378.22 |
| S5 | -1303.5782 | 0.2495 | 59.11 | -95.92 | 303.05 | 43.59 | 160.59 | -256.51 |
| A1 | -1189.5202 | 0.1656 | 48.16 | -287.36 | 247.67 | 34.26 | 121.34 | -408.70 |
| A2 | -1238.7426 | 0.1626 | 48.73 | -329.09 | 248.40 | 20.97 | 112.95 | -442.04 |
| A3 | -1148.5840 | 0.1660 | 48.64 | 77.00 | 255.60 | 19.60 | 115.96 | -38.96 |
| A4 | -1284.1505 | 0.2095 | 55.26 | -223.74 | 283.45 | 39.48 | 145.54 | -369.28 |
| A5 | -1163.5438 | 0.1586 | 46.25 | -274.36 | 235.64 | 33.11 | 114.16 | -388.52 |
| A6 | -1073.3879 | 0.1622 | 46.48 | 125.49 | 245.53 | 34.74 | 120.45 | 5.04 |
| A7 | -1208.9516 | 0.2057 | 52.52 | -168.48 | 270.41 | 44.86 | 140.33 | -308.81 |
| A8 | -1122.6073 | 0.1589 | 46.79 | 90.59 | 243.03 | 15.95 | 105.96 | -15.37 |
| A9 | -1258.1733 | 0.2025 | 53.02 | -208.96 | 268.54 | 38.72 | 135.93 | -344.89 |
| A10 | -1168.0149 | 0.2061 | 53.04 | 197.23 | 278.12 | 38.32 | 141.56 | 55.67 |
| a The scaling factors for ZPE and HT were 0.98 and 0.96, respectively. | ||||||||
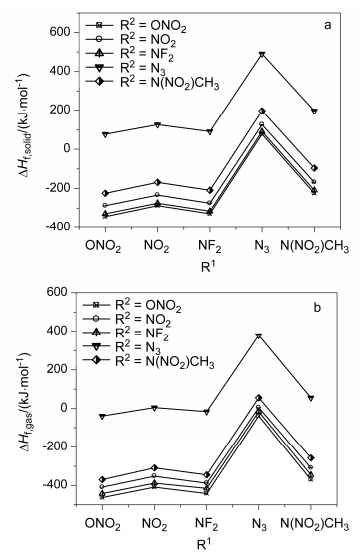
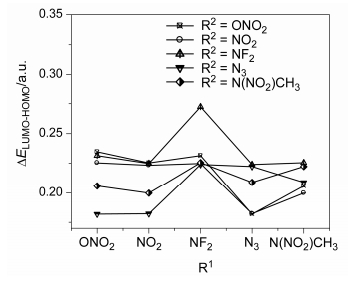
Table 3 presents the calculated highest occupied molecular orbital (HOMO) and lower unoccupied molecular orbital (LUMO) energies and energy gaps (∆ELUMO-HOMO) of the title compounds at B3LYP/6-311++G(d, p) level. Figure 4 shows the effects of different substituents on ∆ELUMO-OMO of the title compounds. For most of the A series, the substitution of NF2 increases the LUMO-HOMO gaps comparing with the corresponding S series except for A2. The symmetric substituted series behave higher energy gaps of LUMO-HOMO than the corresponding asymmetric substituted compounds except for A1, A5, A8 and A9. This indicates that the incorporation of NF2 into NO2, N3 and N(NO2)CH3 substituted compounds and ONO2 into NO2-substituted compounds will decrease the reactivity, respectively. However, the LUMO-HOMO gaps are suggestive of reactivity only as approximation. In all, S3 has the highest value of ∆ELUMO-HOMO among the title compounds.
下载:
导出CSV
| Compd. | EHOMO/a.u. | ELUMO/a.u. | ∆ELUMO-HOMO/a.u. |
| S1 | -0.3346 | -0.0999 | 0.2347 |
| S2 | -0.3281 | -0.1048 | 0.2233 |
| S3 | -0.3229 | -0.0506 | 0.2723 |
| S4 | -0.2777 | -0.0553 | 0.2224 |
| S5 | -0.2917 | -0.0693 | 0.2224 |
| A1 | -0.3266 | -0.1012 | 0.2254 |
| A2 | -0.3259 | -0.0944 | 0.2315 |
| A3 | -0.2799 | -0.0979 | 0.1820 |
| A4 | -0.2984 | -0.0925 | 0.2059 |
| A5 | -0.3264 | -0.1016 | 0.2248 |
| A6 | -0.2816 | -0.0992 | 0.1824 |
| A7 | -0.2984 | -0.0986 | 0.1998 |
| A8 | -0.2802 | -0.0562 | 0.2240 |
| A9 | -0.2973 | -0.0717 | 0.2256 |
| A10 | -0.2780 | -0.0691 | 0.2089 |
Table 4 lists the calculated density, molecular and detonation properties and impact sensitivity of the title compounds at B3PW91/6-31G(d, p) level. Figure 5 presents the effects of different substituents on ρ, D, P and h50 of the title compounds.
下载:
导出CSV
| Compd. | M/V(0.001)/(g•cm-3) | Crystal density/(g•cm-3) | Q/(cal•g-1) | D/(km•s-1) | P/GPa | Vb | h50/cm | ||
| S1 | 1.90 | 23.54 | 1.85 | 1528.4 | 8.92 | 35.89 | 123.46 | 0.179694 | 39.16 |
| S2 | 1.86 | 32.57 | 1.84 | 1458.1 | 8.65 | 33.62 | 155.39 | 0.197494 | 43.25 |
| S3 | 1.99 | 14.39 | 1.91 | 1520.9 | 9.01 | 37.31 | 105.79 | 0.140591 | 29.83 |
| S4 | 1.71 | 18.73 | 1.67 | 1450.2 | 7.88 | 26.29 | 78.34 | 0.220414 | 49.28 |
| S5 | 1.71 | 38.06 | 1.72 | 1366.2 | 7.96 | 27.31 | 84.96 | 0.239974 | 53.96 |
| A1 | 1.88 | 32.27 | 1.86 | 1494.1 | 8.84 | 35.37 | 141.83 | 0.204702 | 45.08 |
| A2 | 1.94 | 23.36 | 1.89 | 1522.1 | 9.01 | 37.02 | 121.41 | 0.181812 | 39.69 |
| A3 | 1.81 | 21.75 | 1.77 | 1495.1 | 8.45 | 31.39 | 102.07 | 0.199145 | 43.99 |
| A4 | 1.79 | 36.15 | 1.79 | 1436.5 | 8.44 | 31.47 | 103.74 | 0.247193 | 55.58 |
| A5 | 1.92 | 32.07 | 1.90 | 1485.6 | 8.91 | 36.35 | 131.11 | 0.21593 | 47.86 |
| A6 | 1.78 | 30.90 | 1.77 | 1446.7 | 8.30 | 30.28 | 107.56 | 0.234407 | 52.47 |
| A7 | 1.77 | 39.38 | 1.78 | 1403.8 | 8.28 | 30.23 | 109.55 | 0.248915 | 55.96 |
| A8 | 1.85 | 16.79 | 1.79 | 1489.5 | 8.45 | 31.55 | 93.43 | 0.180208 | 39.48 |
| A9 | 1.83 | 34.56 | 1.82 | 1430.2 | 8.47 | 32.01 | 93.51 | 0.249796 | 56.28 |
| A10 | 1.71 | 34.83 | 1.71 | 1399.1 | 7.96 | 27.21 | 87.25 | 0.249176 | 56.17 |
| HMXc | 1.91 | 9.10 | 39.60 | 30 | |||||
| a The computed results pertain to 0.001 a.u. surfaces.b The computed results pertain to 0.002 a.u. surfaces. c Calculated values taken from Ref. [44]. | |||||||||
The substitution of NF2 increases and has the highest values of ρ, D and P for almost the entire S and A series of 9 title compounds except for A2. A2 has the same value of D comparing with S3. This indicates that NF2 group is the most useful energetic substituent for improving the calculated density and detonation properties of 3, 3-bis(difluo- roamino)-1, 5-substituted pentane derivatives. The values of h50 are on the range of 29~57 cm. S3 has the smallest value among them, while A9 had the largest one. The N(NO2)CH3 groups obviously improve the impact sensitivity among other mentioned energetic groups. S3 has the similar stability compared with HMX. Besides, S3 has the highest crystal density and detonation properties which could be considered as a promising energetic plasticizer.
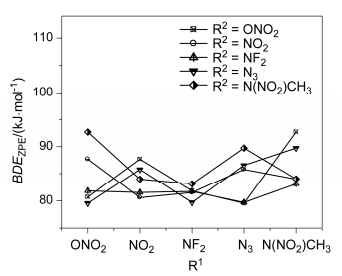
The values of BDEZPE of relatively weak bonds for the title compounds at B3LYP/6-311++G(d, p) level are listed in Table 5. The C—NF2 bond locates in the central position of pentane had the lowest value of BDEZPE among the title compounds, which suggests that the cleavage of central C—NF2 bond would be a possible initial thermal decomposition path for these compounds. Figure 6 shows the effects of different substituents on BDEZPE of central C—NF2 bond. The substitution of ONO2 and N(NO2)CH3 can increase the value of BDEZPE of central C—NF2 bond. And A4 has the highest BDEZPE. This could be caused by the relatively stronger steric hindrance of these two energetic groups. Considering the suggested energy barrier (80 kJ/mol), [45] most of these compounds can be thermal stable to some degree except for A3 and A8.
下载:
导出CSV
| Compd. | BDEZPE/(kJ•mol-1) | ||||||||
| C—NF2a | C—NF2b | C—ONO2 | O—NO2 | C—NO2 | N—NO2 | C—N3 | N—Fa | N—Fb | |
| S1 | 80.67 | 264.69 | 104.48 | 217.29 | |||||
| S2 | 80.58 | 185.04 | 214.79 | ||||||
| S3 | 81.70 | 205.77 | 209.34 | 232.03 | |||||
| S4 | 86.57 | 244.93 | 216.79 | ||||||
| S5 | 84.01 | 129.10 | 216.72 | ||||||
| A1 | 87.71 | 263.33 | 103.38 | 184.83 | 211.32 | ||||
| A2 | 81.84 | 205.65 | 265.33 | 104.07 | 215.55 | 232.09 | |||
| A3 | 79.55 | 265.93 | 104.68 | 236.86 | 217.32 | ||||
| A4 | 92.72 | 267 | 105.23 | 139.93 | 217.10 | ||||
| A5 | 81.54 | 205.33 | 186.52 | 215.32 | 229.65 | ||||
| A6 | 85.80 | 193.69 | 243.11 | 214.92 | |||||
| A7 | 84.00 | 187.41 | 128.83 | 215.58 | |||||
| A8 | 79.68 | 207.41 | 238.03 | 216.49 | 230.32 | ||||
| A9 | 83.11 | 206.91 | 129.00 | 215.57 | 232.20 | ||||
| A10 | 89.76 | 129.86 | 238.12 | 214.72 | |||||
| a Bond located in the 3 position of the title compounds; b bond located in the 1 or 5 position of the title compounds. | |||||||||
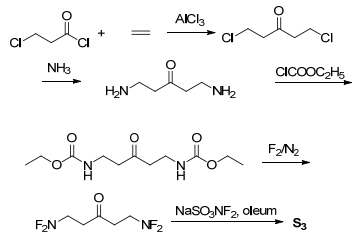
As S3 has the outstanding energy properties, its syntheticroute is speculated via 5 steps (Figure 7). 1, 5-Dichloro- pentan-3-one can be synthesized via the reaction of 3-chloropropionyl chloride, ethylene and aluminum chloride.[46] Then, by the following processes of amination, [47] amidation, [8, 9] fluorination[8, 9] and difluoroamination, [48] S3 could be gained.
The HOFs, energy properties, and thermal stability of a series of 3, 3-bis(difluoroamino)-1, 5-substituted pentane derivatives with different energetic substituents are studied using DFT methods. The azido group significantly increases the HOFs both in gas and solid states. But the difluoroamino group obviously improves the values of crystal density, detonate velocity and detonation pressure. The weakest bond is the C—NF2 bond located in the third position of the title compounds, which indicates that its cleavage could possibly be the initial thermal decomposi tion path. Most of the BDEZPE values are above the suggested energy barrier. Besides, S3 is chosen as a potential energetic plasticizer for its highest detonation properties, lowest electronic reactivity and acceptable impact sensitivity. By using a five-step synthetic route, S3 can be synthesized as a novel difluoroamino energetic plasticizer.
Supporting Information Details of the Kamlet-Jacobs equation are included, especially, the formula derivation of the parameters N,
Chapman, R. D. In Organic Difluoramine Derivatives, Vol. 125, Ed.: Klap tke, T. M., Springer, Berlin, 2007.
Chapman, R. D.; Welker, M. F.; Kreutzberger, C. B. J. Org. Chem. 1998, 63, 1566. doi: 10.1021/jo9718399
Chapman, R. D.; Gilardi, R. D.; Welker, M. F.; Kreutzberger, C. B. J. Org. Chem. 1999, 64, 960. doi: 10.1021/jo9819640
Chapman, R. D.; Groshens, T. J. US 7632943, 2009[Chem. Abstr. 2009, 152, 57346].
Zhang, J.; Oxley, J.; Smith, J.; Bedford, C.; Chapman, R. J. Mass Spectrom. 2000, 35, 841. doi: 10.1002/(ISSN)1096-9888
Chapman, R. D.; Nguyen, B. V. US 6310204, 2001[Chem. Abstr. 2001, 135, 346536].
Axenrod, T.; Guan, X. P.; Sun, J.; Qi, L.; Chapman, R. D.; Gliardi, R. D. Tetrahedron Lett. 2001, 42, 2621. doi: 10.1016/S0040-4039(01)00260-X
Archibald, T. G.; Manser, G. E.; Immoos, J. E. US 5272249, 1993[Chem. Abstr. 1994, 120, 135476].
Archibald, T. G.; Manser, G. E.; Immoos, J. E. US 5420311, 1995[Chem. Abstr. 1994, 120, 135476].
Archibald, T. G.; Manser, G. E. US 5789617, 1998[Chem. Abstr. 1994, 120, 298071].
Adolph, H. G.; Trivedi, N. J. US 6325876, 2001[Chem. Abstr. 2001, 136, 8637].
Li, H.; Pan, R. M.; Wang, W. J.; Zhang, L. Y. Propellants, Explos., Pyrotech. 2014, 39, 819. doi: 10.1002/prep.201400036
Li, H.; Pan, R. M.; Wang, W. J.; Zhang, L. Y. J. Therm. Anal. Calorim. 2014, 118, 189. doi: 10.1007/s10973-014-3985-y
Li, H.; Pan, J. A.; Wang, W. J.; Pan, R. M.; Zhu, W. H. J. Macromol. Sci., Part A:Pure Appl. Chem. 2018, 55, 135. doi: 10.1080/10601325.2017.1387742
Wu, Q.; Zhu, W. H.; Xiao, H. M. J. Mol. Model. 2013, 19, 2945. doi: 10.1007/s00894-013-1825-9
Pan, Y.; Li, J. S.; Cheng, B. B.; Zhu, W. H.; Xiao, H. M. Comput. Theor. Chem. 2012, 992, 110. doi: 10.1016/j.comptc.2012.05.013
Wu, Q.; Pan, Y.; Zhu, W. H.; Xiao, H. M. J. Mol. Model. 2013, 19, 1853. doi: 10.1007/s00894-013-1756-5
Jensen, T. L.; Moxnes, J. F.; Kj nstad, E. F.; Unneberg, E. Cent. Eur. J. Energ. Mater. 2016, 13, 445. doi: 10.22211/cejem/64995
Xiang, D.; Chen, H.; Zhu, W. H.; Xiao, H. M. Can. J. Chem. 2016, 94, 667. doi: 10.1139/cjc-2016-0174
Muthurajan, H.; Sivabalan, R.; Talawar, M. B.; Anniyappan, M.; Venugopalan, S. J. Hazard. Mater. 2006, 133, 30. doi: 10.1016/j.jhazmat.2005.10.009
Chen, Z. X.; Xiao J. M.; Xiao, H. M.; Chiu, Y. N. J. Phys. Chem. A 1999, 103, 8062. doi: 10.1021/jp9903209
Ju, X. H.; Li, Y. M.; Xiao, H. M. J. Phys. Chem. A 2005, 109, 934. doi: 10.1021/jp045071p
Ju, X. H.; Wang, X.; Bei, F. L. J. Comput. Chem. 2005, 26, 1263. doi: 10.1002/(ISSN)1096-987X
Atkins, P. W. Physical Chemistry, Oxford University Press, Oxford, 1982.
Politzer, P.; Murry, J. S.; Grice, M. E.; Salvo, M. De; Miller, E. Mol. Phys. 1997, 91, 923. doi: 10.1080/002689797171030
Politzer, P.; Murry, J. S. Cent. Eur. J. Energ. Mater. 2011, 8, 209.
Byrd, E. F. C.; Rice, B. M. J. Phys. Chem. A 2006, 110, 1005. doi: 10.1021/jp0536192
Kamlet, M. J.; Jacobs, S. T. J. Chem. Phys. 1968, 48, 23. doi: 10.1063/1.1667908
孙业斌, 惠君明, 曹欣茂, 军用混合炸药, 兵器工业出版社, 北京, 1995.Sun, Y. B.; Hui, J. M.; Cao, X. M. Military Use of Blended Explosive, Weapon Industry Press, Beijing, 1995 (in Chinese).
Politzer, P.; Martines, J.; Murry, J. S.; Concha, M. C.; Toro-Labbé, A. Mol. Phys. 2009, 107, 2095. doi: 10.1080/00268970903156306
Pospíšil, M.; Vávra, P.; Concha, M. C.; Murry, J. S.; Politzer, P. J. Mol. Model. 2010, 16, 895. doi: 10.1007/s00894-009-0587-x
Benson, S. W. Thermochemical Kinetic, 2nd ed., Weily Interscience, New York, 1976.
Mills, I.; Cvitas, T.; Homann, K.; Kallay, N.; Kuchitsu, K. Quantities, Units, and Symbols in Physical Chemistry, Blackwell Scientific Publications, Oxford, 1988.
Blanksby, S. J.; Ellison, G. B. Acc. Chem. Res. 2003, 36, 255. doi: 10.1021/ar020230d
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010.
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B:Condens. Matter Mater. Phys. 1988, 37, 785. doi: 10.1103/PhysRevB.37.785
Frisch, M. J.; Pople, J. A.; Binkley, J. S. J. Chem. Phys. 1984, 80, 3265. doi: 10.1063/1.447079
Lu, T., and Chen, F. J. Comput. Chem. 2012, 33, 580. doi: 10.1002/jcc.v33.5
Dean, J. A. LANGE's Handbook of Chemistry, 13th ed., Mc Graw-Hill Book Co., New York, 1985.
Dean, J. A. LANGE's Handbook of Chemistry, 15th ed., Mc Graw-Hill Book Co., New York, 1999.
Joo, Y. H.; Shreeve, J. M. Angew. Chem., Int. Ed. 2009, 48, 564. doi: 10.1002/anie.v48:3
Ghule, V. D.; Sarangapani, R.; Jadhav, P. M.; Pandey, R. K. J. Mol. Model. 2011, 17, 2927. doi: 10.1007/s00894-011-0959-x
Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502. doi: 10.1021/jp960976r
Shen, C.; Wang, P. C.; Lu, M. J. Phys. Chem. A 2015, 119, 8250. doi: 10.1021/acs.jpca.5b04969
Chung, G.; Schmidt, M. W.; Gordon, M. S. J. Phys. Chem. A 2000, 104, 5647. doi: 10.1021/jp0004361
Owen, G. R.; Reese, C. B. J. Chem. Soc. C 1970, 17, 2401.
Kenji, H.; Tadashi, M.; Shaoji, S. JP 2007-070270, 2007[Chem. Abstr. 2007, 146, 358683].
Haiges, R.; Wager, R.; Boatz, J. A.; Yousufuddin, M.; Etzkorn, M.; Prakash, G. K.; Christe, K. O.; Chapman, R. D.; Welker, M. F.; Kreutzberger. C. B. Angew. Chem., Int. Ed. 2006, 45, 5179. doi: 10.1002/(ISSN)1521-3773

Figure 1 Molecular frameworks of 3, 3-bis(difluoroamino)- 1, 5-substituted pentane derivatives

Figure 3 Effects of different substituents on ∆Hf, gas (a) and ∆Hf, solid (b) of the title compounds

Figure 5 Effects of different substituents on ρ (a), D (b), P (c) and h50 (d) of the title compounds
Table 1. Calculated total energy (E0), zero-point energies (ZPE), thermal corrections (HT) and heat of formation (HOFs) of the reference compounds at B3LYP/6-311++G(d, p) level
| Compd. | E0/a.u. | ZPEa/a.u. | HTa/(kJ•mol-1) | HOF/(kJ•mol-1) |
| H4 | -40.4894 | 0.04365 | 9.61 | -74.60b |
| C5H12 | -197.6702 | 0.15650 | 20.30 | -146.90b |
| CH3NF2 | -294.2638 | 0.04564 | 13.14 | -115.23d |
| CH3ONO2 | -320.2390 | 0.05292 | 15.00 | -124.40b |
| CH3NO2 | -245.0420 | 0.04864 | 13.35 | -74.30b |
| CH3N3 | -204.1033 | 0.04908 | 13.64 | 296.54e |
| (CH3)2NNO2 | -339.6683 | 0.09274 | 19.26 | -0.84c |
| a The scaling factors for ZPE and HT were 0.98 and 0.96, respectively.[43] bExperimental values taken from Ref. [40]. cExperimental values taken from Ref. [39]. d Calculated values taken from Ref. [22]; e Calculated values taken from Ref. [23]. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Calculated E0, ZPE, HT, molecular properties, heat of sublimation and HOFs of the title compounds at B3LYP/6-311++G(d, p) level
| Compd. | E0/a.u. | ZPEa/a.u. | HT/(kJ•mol-1) | ∆Hf, gas/(kJ•mol-1) | A/A2 | ∆Hsub/(kJ•mol-1) | ∆Hf, solid/(kJ•mol-1) | |
| S1 | -1264.7195 | 0.1697 | 50.21 | -343.57 | 258.68 | 21.85 | 119.43 | -463.00 |
| S2 | -1114.3217 | 0.1617 | 45.92 | -232.92 | 237.41 | 39.48 | 118.75 | -351.67 |
| S3 | -1212.7655 | 0.1555 | 46.70 | -314.63 | 233.80 | 14.86 | 100.09 | -414.72 |
| S4 | -1032.4513 | 0.1625 | 47.05 | 490.73 | 253.08 | 17.10 | 112.51 | 378.22 |
| S5 | -1303.5782 | 0.2495 | 59.11 | -95.92 | 303.05 | 43.59 | 160.59 | -256.51 |
| A1 | -1189.5202 | 0.1656 | 48.16 | -287.36 | 247.67 | 34.26 | 121.34 | -408.70 |
| A2 | -1238.7426 | 0.1626 | 48.73 | -329.09 | 248.40 | 20.97 | 112.95 | -442.04 |
| A3 | -1148.5840 | 0.1660 | 48.64 | 77.00 | 255.60 | 19.60 | 115.96 | -38.96 |
| A4 | -1284.1505 | 0.2095 | 55.26 | -223.74 | 283.45 | 39.48 | 145.54 | -369.28 |
| A5 | -1163.5438 | 0.1586 | 46.25 | -274.36 | 235.64 | 33.11 | 114.16 | -388.52 |
| A6 | -1073.3879 | 0.1622 | 46.48 | 125.49 | 245.53 | 34.74 | 120.45 | 5.04 |
| A7 | -1208.9516 | 0.2057 | 52.52 | -168.48 | 270.41 | 44.86 | 140.33 | -308.81 |
| A8 | -1122.6073 | 0.1589 | 46.79 | 90.59 | 243.03 | 15.95 | 105.96 | -15.37 |
| A9 | -1258.1733 | 0.2025 | 53.02 | -208.96 | 268.54 | 38.72 | 135.93 | -344.89 |
| A10 | -1168.0149 | 0.2061 | 53.04 | 197.23 | 278.12 | 38.32 | 141.56 | 55.67 |
| a The scaling factors for ZPE and HT were 0.98 and 0.96, respectively. | ||||||||
下载: 导出CSV
Table 3. Calculated HOMO and LUMO energies and energy gaps ∆ELUMO-HOMO of the title compounds at B3LYP/6-311++G(d, p) level
| Compd. | EHOMO/a.u. | ELUMO/a.u. | ∆ELUMO-HOMO/a.u. |
| S1 | -0.3346 | -0.0999 | 0.2347 |
| S2 | -0.3281 | -0.1048 | 0.2233 |
| S3 | -0.3229 | -0.0506 | 0.2723 |
| S4 | -0.2777 | -0.0553 | 0.2224 |
| S5 | -0.2917 | -0.0693 | 0.2224 |
| A1 | -0.3266 | -0.1012 | 0.2254 |
| A2 | -0.3259 | -0.0944 | 0.2315 |
| A3 | -0.2799 | -0.0979 | 0.1820 |
| A4 | -0.2984 | -0.0925 | 0.2059 |
| A5 | -0.3264 | -0.1016 | 0.2248 |
| A6 | -0.2816 | -0.0992 | 0.1824 |
| A7 | -0.2984 | -0.0986 | 0.1998 |
| A8 | -0.2802 | -0.0562 | 0.2240 |
| A9 | -0.2973 | -0.0717 | 0.2256 |
| A10 | -0.2780 | -0.0691 | 0.2089 |
下载: 导出CSV
Table 4. Table 4 Calculated uncorrected density [M/V(0.001)], corrected density (crystal density), detonation velocity (D), detonation pressure (P), molecular properties and impact sensitivity (h50) of the title compounds at B3PW91/6-31G(d, p) level
| Compd. | M/V(0.001)/(g•cm-3) | Crystal density/(g•cm-3) | Q/(cal•g-1) | D/(km•s-1) | P/GPa | Vb | h50/cm | ||
| S1 | 1.90 | 23.54 | 1.85 | 1528.4 | 8.92 | 35.89 | 123.46 | 0.179694 | 39.16 |
| S2 | 1.86 | 32.57 | 1.84 | 1458.1 | 8.65 | 33.62 | 155.39 | 0.197494 | 43.25 |
| S3 | 1.99 | 14.39 | 1.91 | 1520.9 | 9.01 | 37.31 | 105.79 | 0.140591 | 29.83 |
| S4 | 1.71 | 18.73 | 1.67 | 1450.2 | 7.88 | 26.29 | 78.34 | 0.220414 | 49.28 |
| S5 | 1.71 | 38.06 | 1.72 | 1366.2 | 7.96 | 27.31 | 84.96 | 0.239974 | 53.96 |
| A1 | 1.88 | 32.27 | 1.86 | 1494.1 | 8.84 | 35.37 | 141.83 | 0.204702 | 45.08 |
| A2 | 1.94 | 23.36 | 1.89 | 1522.1 | 9.01 | 37.02 | 121.41 | 0.181812 | 39.69 |
| A3 | 1.81 | 21.75 | 1.77 | 1495.1 | 8.45 | 31.39 | 102.07 | 0.199145 | 43.99 |
| A4 | 1.79 | 36.15 | 1.79 | 1436.5 | 8.44 | 31.47 | 103.74 | 0.247193 | 55.58 |
| A5 | 1.92 | 32.07 | 1.90 | 1485.6 | 8.91 | 36.35 | 131.11 | 0.21593 | 47.86 |
| A6 | 1.78 | 30.90 | 1.77 | 1446.7 | 8.30 | 30.28 | 107.56 | 0.234407 | 52.47 |
| A7 | 1.77 | 39.38 | 1.78 | 1403.8 | 8.28 | 30.23 | 109.55 | 0.248915 | 55.96 |
| A8 | 1.85 | 16.79 | 1.79 | 1489.5 | 8.45 | 31.55 | 93.43 | 0.180208 | 39.48 |
| A9 | 1.83 | 34.56 | 1.82 | 1430.2 | 8.47 | 32.01 | 93.51 | 0.249796 | 56.28 |
| A10 | 1.71 | 34.83 | 1.71 | 1399.1 | 7.96 | 27.21 | 87.25 | 0.249176 | 56.17 |
| HMXc | 1.91 | 9.10 | 39.60 | 30 | |||||
| a The computed results pertain to 0.001 a.u. surfaces.b The computed results pertain to 0.002 a.u. surfaces. c Calculated values taken from Ref. [44]. | |||||||||
下载: 导出CSV
Table 5. Bond dissociation energies (BDE) of the relatively weak bonds for the title compounds at B3LYP/6-311++G(d, p) level
| Compd. | BDEZPE/(kJ•mol-1) | ||||||||
| C—NF2a | C—NF2b | C—ONO2 | O—NO2 | C—NO2 | N—NO2 | C—N3 | N—Fa | N—Fb | |
| S1 | 80.67 | 264.69 | 104.48 | 217.29 | |||||
| S2 | 80.58 | 185.04 | 214.79 | ||||||
| S3 | 81.70 | 205.77 | 209.34 | 232.03 | |||||
| S4 | 86.57 | 244.93 | 216.79 | ||||||
| S5 | 84.01 | 129.10 | 216.72 | ||||||
| A1 | 87.71 | 263.33 | 103.38 | 184.83 | 211.32 | ||||
| A2 | 81.84 | 205.65 | 265.33 | 104.07 | 215.55 | 232.09 | |||
| A3 | 79.55 | 265.93 | 104.68 | 236.86 | 217.32 | ||||
| A4 | 92.72 | 267 | 105.23 | 139.93 | 217.10 | ||||
| A5 | 81.54 | 205.33 | 186.52 | 215.32 | 229.65 | ||||
| A6 | 85.80 | 193.69 | 243.11 | 214.92 | |||||
| A7 | 84.00 | 187.41 | 128.83 | 215.58 | |||||
| A8 | 79.68 | 207.41 | 238.03 | 216.49 | 230.32 | ||||
| A9 | 83.11 | 206.91 | 129.00 | 215.57 | 232.20 | ||||
| A10 | 89.76 | 129.86 | 238.12 | 214.72 | |||||
| a Bond located in the 3 position of the title compounds; b bond located in the 1 or 5 position of the title compounds. | |||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们