Table 1.
Representative results for optimization of Ni-catalyzed difluoromethylation of 2a with BrCF2H (1)a
Citation:
Gao Xing, He Xu, Zhang Xingang. Nickel-Catalyzed Difluoromethylation of (Hetero)aryl Bromides with BrCF2H[J]. Chinese Journal of Organic Chemistry,
2019, 39(1): 215-222.
doi:
10.6023/cjoc201808014

镍催化下一溴二氟甲烷对(杂)芳基溴代物的二氟甲基化反应
摘要:
二氟甲基取代的(杂)芳烃化合物由于二氟甲基的独特性质越来越受到了化学家和药物学家的广泛关注.在过去的几年中,大量合成该类化合物的方法不断被发展,但这些方法中大多使用了价格昂贵的需要多步合成的二氟甲基化试剂,从而制约了其广泛应用.因此,发展以廉价易得的二氟甲基化试剂为氟源制备二氟甲基(杂)芳烃化合物的方法是十分必要的.以廉价易得的溴二氟甲烷(BrCF2H)为二氟甲基化试剂,发展了镍催化下(杂)芳基溴代物与BrCF2H的偶联反应.该反应高效温和,底物普适性好,官能团兼容性优秀,并且可以克量级合成,为合成二氟甲基(杂)芳烃化合物提供了一种高效简洁、廉价的方法.初步机理研究表明,该反应经历了一种镍催化的还原偶联历程.
English
Nickel-Catalyzed Difluoromethylation of (Hetero)aryl Bromides with BrCF2H
Abstract:
A nickel-catalyzed direct difluoromethylation of (hetero)aryl bromides with bromodifluoromethane (BrCF2H) is described. This reaction features high efficiency, broad substrate scope and high functional group tolerance, providing a cost-efficient and straightforward route for applications in medicinal chemistry. Preliminary mechanistic studies reveal that a nickel-based, reductive cross-coupling catalytic cycle is involved in the reaction.
-
Key words:
- aryl bromide
- / bromodifluoromethane
- / difluoromethylation
- / nickel
- / reductive cross-coupling
-
1. Introduction
Difluoromethylated (hetero)arenes have gained increasing attention because of the unique properties of difluoromethyl group (CF2H) that can function as a lipophilic hydrogen bond donor, and also as a bioisostere of hydroxyl and thiol groups.[1] Hence, it is of great interest to develop new and efficient methods to synthesize difluoromethylated (hetero)arenes. Transition-metal-catalyzed direct difluromethylation of (hetero)aromatics has emerged as one of the most efficient strategies to access difluoromethylated (hetero)arenes.[2] However, most of the difluoromethylation reagents used in these reported methods are expensive and additional step(s) are required to synthesize from fluoroalkyl halides, [2c, 3] thus limiting their widespread applications.
In this regard, recently, we developed a palladium-catalyzed difluoromethylation of arylboronic acids with abundant and inexpensive industrial material chlorodifluoromethane (ClCF2H) via a metal-difluorocarbene coupling (MeDiC), [4] which represents the first example of catalytic difluoromethylation from ClCF2H.[5] Later on, to replace the palladium with a base metal as a catalyst, we[6] developed a nickel-catalyzed reductive cross-coupling between ClCF2H and (hetero)aryl chlorides/bromides. Despite of the importance of these methods, the developing new methods to access valuable difluoromethylated (hetero)arenes remain highly desirable. Bromodifluoromethane (BrCF2H) is a simple and readily available difluoromethyl source.[7] Very recently, we[8] and others[9] reported a nickel-catalyzed cross-coupling of BrCF2H with arylboronic acids, independently. However, the nickel-catalyzed reductive cross-coupling of BrCF2H with (hetero)aryl halides has not been reported so far. To continue our research on catalytic fluoroalkylation reactions with inexpensive and readily available fluoroalkyl halides as fluoroalkyl sources, [10] herein, we disclose a nickel-catalyzed reductive cross- coupling of widely accessible (hetero)aryl bromides with BrCF2H. The reaction proceeds under mild reaction conditions with broad substrate scope and excellent functional group tolerance without preformation of arylmetals.
2. Results and discussion
Initially, 4-bromo-1, 1'-biphenyl (2a) was chosen as a coupling partner for this nickel-catalyzed difluoromethylation reaction (Table 1). However, only 16% yield of 3a was provided when the reaction was carried out with BrCF2H (1, 1.0 equiv.) and 2a (2.5 equiv.) in the presence of NiBr2•Diglyme (5 mol%) and bypyridyl (bpy) (5 mol%) in 1, 3-dimethyl-3, 4, 5, 6-tetrahydro-2(1H)-pyrimidinone (DMPU) at 80 ℃ by employing Zn as a reductant (Entry 1). The low yield of 3a is because of the formation of hydrodebrominated compound CF2H2 (4, 10% yield) and some other uncertain defluorinated by-products. To suppress these side reactions, a series of reaction parameters, such as ligand, solvent, and additive were examined (Entries 2~7). Among the tested ligands and solvents, the bpy ligand and DMPU were still the best choice. Other diamine ligands, such as 1, 10-phenanthroline (phen), 4, 4'-ditBu-bpy, even the electron-rich ligand 4, 4'-diMeO-bpy, all provided lower yields. To our delight, the use of KI (0.4 equiv.)[11] as an additive could afford 3a in 66% yield albeit formation of 5% yield of CF2H2 (Entry 6). The reaction was sensitive to the nickel catalysts (Entries 7~11) and Ni(PPh3)2Br2 showed the highest reactivity (Entry 6). Finally, the optimum reaction conditions were identified by decreasing the reaction temperature to 60 ℃ with utility of 1.5 equiv. of 2a and 10 mol% Ni(PPh3)2Br2/bpy, providing compound 3a in 91% yield upon isolation (Entry 13). No product 3a was observed without nickel or bpy, thus demonstrating the essential role of Ni/bpy in promotion of the reaction (Entries 14 and 15).
Table 1

 下载:
导出CSV
下载:
导出CSV

Entry [Ni] Zn/equiv. Additive (equiv.) Yieldb/% of 3a/4/1 1 NiBr2•Diglyme 1.2 — 16/10/36 2 NiBr2•Diglyme 1.2 LiI (0.2) 24/Trace/44 3 NiBr2•Diglyme 1.2 NaI (0.2) 30/12/21 4 NiBr2•Diglyme 1.2 KI (0.2) 45/Trace/25 5 NiBr2•Diglyme 2.0 KI (0.2) 62/Trace/7 6 NiBr2•Diglyme 2.0 KI (0.4) 66/5/0 7 Ni(PPh3)2Br2 2.0 KI (0.4) 73/5/0 8 Ni(PPh3)2Cl2 2.0 KI (0.4) 60/12/0 9 Ni(dppe)Cl2 2.0 KI (0.4) 27/12/0 10 NiCl2•DME 2.0 KI (0.4) 19/Trace/34 11 Ni(cod)2 2.0 KI (0.4) 0/8/45 12c Ni(PPh3)2Br2 2.0 KI (0.4) 78/4/0 13c, d Ni(PPh3)2Br2 2.0 KI (0.4) 90 (91)/Trace/0 14c, d — 2.0 KI (0.4) 0/9/0 15c, d, e Ni(PPh3)2Br2 2.0 KI (0.4) 0/8/10 a Reaction conditions (unless otherwise specified): 1 (0.3 mmol, 1.0 equiv.), 2a (0.75 mmol, 2.5 equiv.), DMPU (2 mL), 6 h. b Determined by 19F NMR using fluorobenzene as an internal standard and number in parenthesis is isolated yield. c Ni(PPh3)2Br2 (10 mol%), bpy (10 mol%), 60 ℃, 6 h. d 1 (0.3 mmol, 1.0 equiv.), 2a (0.45 mmol, 1.5 equiv.), DMPU (1.5 mL). e The reaction was run in the absence of bpy. With the viable reaction conditions in hand, a variety of aryl bromides were examined (Table 2). Generally, arylbromides bearing both electron-donating and electron-withdrawing substituents all showed good reactivity towards BrCF2H, providing the corresponding difluoromethylated arenes in good to high yields (3c~3i). The reaction showed high functional group compatibility. Many important functional groups, including alkoxycarbonyl, enolizable ketone silyl and cyano are compatible with the reaction conditions (3f~3k), even towards alcohol (3l) and arylboronate (3m), thus demonstrating the advantage of present nickel-catalyzed process. Importantly, heteroaryl bromides were also amenable to the reaction. Pyridine-, quinoline-, isoquinoline-, benzothiazole-, and benzooxazole-containing substrates all underwent the reaction smoothly, leading to the corresponding difluoromethylated heteroarenes in good yields (3p~3w). Gram-scale synthesis of difluoromethylated quinoline 3t also proceeded efficiently with comparable yield (64%), thus demonstrating the reliability and practicability of this method. Furthermore, the utility of this method can also be demonstrated by the late-stage difluoromethylation of estrone- and ibuprofen-derived aryl bromides (3x and 3y), providing potential opportunities for discovering new interesting bioactive molecules.
Table 2
Table 2. Ni-catalyzed reductive cross-coupling of bromodifluoromethane (1) with aryl bromides (2)a下载:
导出CSV



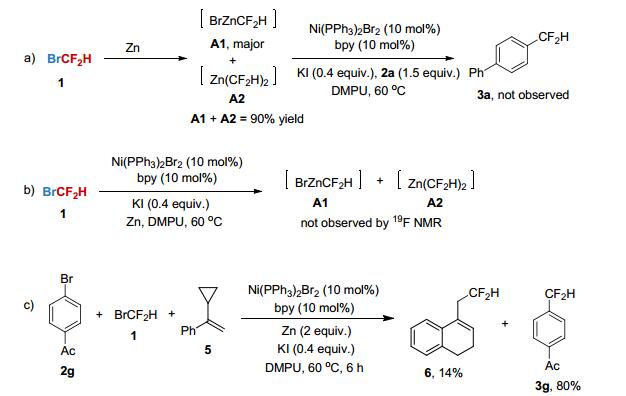
aReaction conditions (unless otherwise specified): 2 (0.9 mmol, 1.5 equiv.), 1 (0.6 mmol, 1.0 equiv.), DMPU (3.0 mL), 60 ℃, 6 h. Yields of isolated products are given. bNMR yield determined by 19F NMR. c2 (2.0 equiv.), 1 (0.6 mmol, 1.0 equiv.), Ni(PPh3)Br2 (5 mol%), 4, 4'-ditBu-bpy (5 mol%), 3Å MS (200 mg), 12 h. dGram-scale synthesis and reaction run for 12 h. e2 (1.5 equiv.), 6 (0.3 mmol, 1.0 equiv.), Ni(PPh3)Br2 (5 mol%), 4, 4'-ditBu-bpy (5 mol%), 3Å MS (400 mg), 12 h. To gain some mechanistic insights into the current nickel-catalyzed process, several experiments were conducted. Firstly, the reaction of BrCF2H with Zn in DMPU was performed at 60 ℃ (Scheme 1a). It was found that the difluoromethyl zinc species [BrZnCF2H] (A1) and [Zn(CF2H)2] (A2) could be generated in a high yield (A1+A2=90% yield determined by 19F NMR)[12] under this reaction condition. However, no desired product 3a was observed when the mixture of A1 and A2 was treated with aryl bromide 2a in the presence of Ni(PPh3)2Br2 and bpy (Scheme 1a). In addition, the use of 19F NMR to monitor the reaction showed that A1 and A2 were not generated under standard reaction conditions (Scheme 1b). Thus, these results rule out the pathway that the production of difluoro-methylated arenes is originated from the cross-coupling of difluoroalkyl zinc species with aryl bromides. Secondly, the radical clock experiment by employing α-cyclopropyl- styrene 5 as a probe[6] showed that a ring-expanded product 6 was obtained in 14% yield, demonstrating that a difluoromethyl radical exists in the reaction (Scheme 1c).
Scheme 1
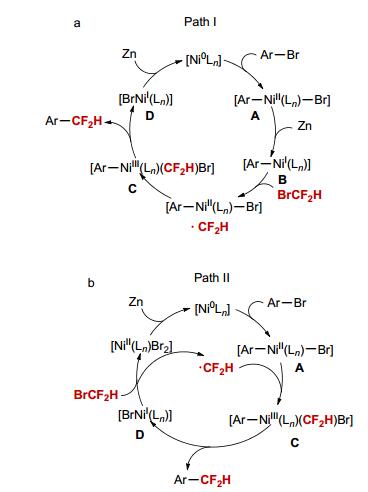
On the basis of these results and previous reports, [6, 13] two possible pathways were proposed for the current nickel-catalyzed process (Scheme 2). Path Ⅰ: The reaction begins the reaction of Ni(0) with aryl bromide to generate Ni(Ⅱ) complex [(Ar)NiI(Ln)Br] (A), which is subsequently reduced by Zn to give Ni(I) complex [(Ar)NiI(Ln)] (B) (Scheme 2a). B reacts with BrCF2H via a radical cage rebound process to produce the key intermediate [(Ar)NiIII-(Ln)(CF2H)Br] (C). Finally, C undergoes reductive elimination to provide the difluoromethylated arene and [BrNiI(Ln)] (D). Reduction of D by Zn regenerates the [Ni0(Ln)] species. Path Ⅱ: The difluoromethyl radical generated between [BrNiI(Ln)] (D) and BrCF2H diffuses to the solution to react with A to give the key intermediate C (Scheme 2b). Subsequently, reductive elimination of C provides the difluoromethylated arene. Finally, the resulting nickel(Ⅱ) complex [NiI(Ln)Br2] is reduced by Zn to regenerate [Ni0(Ln)].
Scheme 2
3. Conclusions
In conclusion, we have developed a nickel-catalyzed direct difluoromethylation of (hetero)aryl bromides with simple and readily available BrCF2H via a reductive cross-coupling. The reaction proceeds under mild reaction conditions with high efficiency and good functional groups tolerance, providing a cost-efficient and straightforward route for the synthesis of difluoromethylated (hetero)arenes that are of great interest in medicinal chemistry.
4. Experimental section
4.1 General information
1H NMR and 13C NMR spectra were recorded on an Agilent MR 400 and Agilent MR 500 spectrometer. 19F NMR was recorded on an Agilent MR 400 spectrometer (CFCl3 as an external standard and low field is positive). NMR yield was determined by 19F NMR using fluorobenzene as an internal standard before working up the reaction.
All reagents were used as received from commercial sources, unless otherwise stated, or prepared as described in the literature. 1, 3-dimethyl-3, 4, 5, 6-tetrahydro-2(1H)-pyrimi- dinone (DMPU) was purchased from TCI and distilled from calcium hydride for twice before use.
4.2 Preparation of BrCF2H stock solution
1, 4-Dioxane (15 mL) was added to a Schlenk tube under argon. BrCF2H gas was then bubbled through the 1, 4-dioxane until the total volume of the solution reach the maximum. The concentration of the BrCF2H (3 mol•L-1) stock solution was determined by 19F NMR spectrum using fluorobenzene as an internal standard.
4.3 General procedure for Ni-catalyzed cross-coup- ling of 1 with 2
To a 25 mL of Schlenk tube were added (hetero)arylbromide 2 (0.9 mmol, 1.5 equiv.), Ni(PPh3)2Br2 (10 mol%), bpy (10 mol%), KI (0.4 equiv.), and Zn (2.0 equiv.) under air. The mixture was then evacuated and backfilled with argon for three times. DMPU (3 mL) and BrCF2H (2.5 mol•L-1 in 1, 4-dioxane, 240 μL, 0.6 mmol, 1.0 equiv.) were added subsequently. The Schlenk tube was screw capped and put into a preheated oil bath (60 ℃). After stirring for 6 h, the reaction mixture was cooled to room temperature, diluted with ethyl acetate, and filtered through a pad of celite. The filtrate was concentrated, and the residue was washed with 10 mL of water, and extracted with organic solvent [V(petroleum ether):V(EtOAc)=20:1] for three times. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated. The residue was purified with silica gel chromatography to give product 3.
4-(Difluoromethyl)-1, 1'-biphenyl (3a):[4] The product (111 mg, 91% yield) was purified with silica gel chromatography (petroleum ether) as a white solid. 1H NMR (400 MHz, CDCl3) δ: 7.72~7.68 (m, 2H), 7.64~7.59 (m, 4H), 7.52~7.47 (m, 2H), 7.44~7.49 (m, 1H), 6.71 (t, J=56.4 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -110.3 (d, J=56.5 Hz, 2F); 13C NMR (125.7 MHz, CDCl3) δ: 143.7 (t, J=2.0 Hz), 140.2, 133.2 (t, J=22.4 Hz), 128.9, 127.9, 127.4, 127.2, 126.0 (t, J=6.0 Hz), 114.7 (t, J=238.5 Hz).
1-(tert-Butyl)-4-(difluoromethyl)benzene (3b):[4] The product (74% yield determined by 19F NMR) was purified with silica gel chromatography (petroleum ether) as a colorless oil. 1H NMR (400 MHz, CDCl ) δ: 7.50~7.44 (m, 4H), 6.63 (t, J=56.8 Hz, 1H), 1.35 (s, 9H); 19F NMR (376 MHz, CDCl3) δ: -109.9 (d, J=56.6 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 154.0 (t, J=2.0 Hz), 131.5 (t, J=22.4 Hz), 125.6, 125.3 (t, J=6.0 Hz), 114.9 (t, J=237.9 Hz), 34.8, 31.2.
1-(Difluoromethyl)-4-methoxybenzene (3c):[4] The product (55% yield determined by 19F NMR) as colorless oil was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=100:1). 1H NMR (400 MHz, CDCl3) δ: 7.44 (d, J=8.6 Hz, 2H), 6.96 (d, J=8.8 Hz, 2H), 6.60 (t, J=56.7 Hz, 1H), 3.84 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -108.3 (d, J=56.8 Hz, 2F); 13C NMR (125.7 MHz, CDCl3) δ: 161.33, 127.1 (t, J=6.0 Hz), 126.74 (t, J=22.7 Hz), 114.9 (t, J=237.3 Hz), 114.0, 55.3.
1-(Difluoromethyl)-3-methoxybenzene (3d):[4] The product (83% yield, determined by 19F NMR) was characterized by 19F NMR and GC-MS analysis. 19F NMR (376 MHz, CDCl3) δ: -111.3 (d, J=37.6 Hz, 2F); GC-MS m/z (%): 158.0 (M+).
1-(Difluoromethyl)-3, 5-dimethoxybenzene (3e):[4] The product (63 mg, 56% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=30:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 6.68~6.62 (m, 2H), 6.56 (t, J=56.4 Hz, 1H), 6.56~6.52 (m, 1H), 3.82 (s, 6H); 19F NMR (376 MHz, CDCl3) δ: -110.9 (d, J=56.4 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 161.0, 136.3 (t, J=22.4 Hz), 114.5 (t, J=239.4 Hz), 103.4 (t, J=6.3 Hz), 102.6 (t, J=1.8 Hz), 55.5.
Ethyl 4-(difluoromethyl)benzoate (3f):[4] The product (114 mg, 95% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=10:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J=8.2 Hz, 2H), 7.58 (d, J=8.1 Hz, 2H), 6.69 (t, J=56.1 Hz, 1H), 4.40 (q, J=7.1 Hz, 2H), 1.41 (t, J=7.1 Hz, 3H); 19F NMR (376 MHz, CDCl3) δ: -112.2 (d, J=56.1 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 165.7, 138.3 (t, J=22.4 Hz), 132.7 (t, J=1.8 Hz), 129. 9, 125.6 (t, J=6.1 Hz), 114.0 (t, J=239.7 Hz), 61.3, 14.3.
1-(4-(Difluoromethyl)phenyl)ethanone (3g):[4] The product (94 mg, 92% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=10:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.04 (d, J=8.0 Hz, 2H), 7.61 (d, J=7.9 Hz, 2H), 6.69 (t, J=56.1 Hz, 1H), 2.64 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -112.3 (d, J=56.1 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 197.3, 138.8 (t, J=1.8 Hz), 138.5 (t, J=22.4 Hz), 128.6, 125.9 (t, J=6.0 Hz), 113.9 (t, J=239.8 Hz), 26.8.
1-(3-(Difluoromethyl)phenyl)ethanone (3h):[4] The product (82 mg, 80% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=10:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.06 (s, 1H), 8.04 (d, J=8.2 Hz, 1H), 7.69 (d, J=7.7 Hz, 1H), 7.55 (t, J=7.7 Hz, 1H), 6.68 (t, J=56.2 Hz, 1H), 2.61 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -111.2 (d, J=56.2 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 197.0, 137.4, 134.8 (t, J=22.8 Hz), 130.4 (t, J=1.7 Hz), 129.9 (t, J=5.8 Hz), 129.1, 125.4 (t, J=6.2 Hz), 114.1 (t, J=239.4 Hz), 26.5.
1-(Difluoromethyl)-4-(trifluoromethyl)benzene (3i):[4] The product (77% yield, determined by 19F NMR) was characterized by 19F NMR and GC-MS analysis. 19F NMR (376 MHz, CDCl3) δ: -113.6 (d, J=37.6 Hz, 2F), -64.0 (s, 3F); GC-MS m/z (%): 196.0 (M+).
(4-(Difluoromethyl)phenyl)trimethylsilane (3j):[4] The product (90 mg, 75% yield) was purified with silica gel chromatography (petroleum ether) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.61 (d, J=8.1 Hz, 2H), 7.48 (d, J=7.8 Hz, 2H), 6.63 (t, J=56.4 Hz, 1H), 0.28 (s, 9H); 19F NMR (376 MHz, CDCl3) δ: -110.9 (d, J=56.5 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 144.0 (t, J=1.7 Hz), 134.6 (t, J=22.1 Hz), 133.6, 124.7 (t, J=6.0 Hz), 114.8 (t, J=238.5 Hz), -1.3.
2-(4-(Difluoromethyl)phenyl)acetonitrile (3k): The product (73 mg, 73% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=20:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.52 (d, J=8.0 Hz, 2H), 7.42 (d, J=8.0 Hz, 2H), 6.65 (t, J=56.3 Hz, 1H), 3.79 (s, 2H); 19F NMR (376 MHz, CDCl3) δ: -111.0 (d, J=56.3 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 134.2 (t, J=22.7 Hz), 132.6 (t, J=2.0 Hz), 128.2, 126.3 (t, J=6.1 Hz), 117.3, 114.1 (t, J=238.9 Hz), 23.4; IR (film) νmax: 2965, 2253, 1621, 1425, 1328 cm-1; MS (EI) m/z (%): 167 (M+), 116 (100); HRMS calcd for C9H7NF2 167.0547, found 167.0552.
(4-(Difluoromethyl)phenyl)methanol (3l):[4] The product (57 mg, 60% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=10:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.48 (d, J=8.1 Hz, 2H), 7.41 (d, J=8.0 Hz, 2H), 6.63 (t, J=56.5 Hz, 1H), 4.69 (s, 2H), 2.48 (s, 1H); 19F NMR (376 MHz, C6D6) δ: -111.6 (d, J=56.5 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 143.5 (t, J=1.9 Hz), 133.5 (t, J=22.4 Hz), 126.9, 125.7 (t, J=6.1 Hz), 114.6 (t, J=238.4 Hz), 64.5.
2-(4-(Difluoromethyl)phenyl)-4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane (3m): The product (108 mg, 71% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=100:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.89 (d, J=7.7 Hz, 2H), 7.50 (d, J=7.8 Hz, 2H), 6.65 (t, J=56.4 Hz, 1H), 1.35 (s, 12H); 19F NMR (282 MHz, CDCl3) δ: -111.9 (d, J=56.4 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 136.8 (t, J=22.1 Hz), 135.0, 126.0 (t, J=6.1 Hz), 124.7 (t, J=6.0 Hz), 114.7 (t, J=239.0 Hz), 84.1, 24.9; IR (film) νmax: 2984, 1742, 1374 cm-1; MS (EI) m/z (%): 254 (M+), 239 (100); HRMS calcd. for C13H1610BO2F2 (M-H+) 252.1248, found 252.1247.
1-(Difluoromethyl)naphthalene (3n):[4] The product (64 mg, 60% yield) was purified with silica gel chromatography (Petroleum ether) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ: 8.20 (d, J=8.0 Hz, 1H), 7.98 (d, J=8.3 Hz, 1H), 7.93 (d, J=8.0 Hz, 1H), 7.71 (d, J=7.1 Hz, 1H), 7.67~7.44 (m, 3H), 7.15 (t, J=55.1 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -110.9 (d, J=55.1 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 133.7, 131.5 (t, J=1.8 Hz), 129.7 (t, J=2.9 Hz), 129.5 (t, J=20.9 Hz), 128.7, 127.1, 126.3, 124.8 (t, J=8.7 Hz), 124.6, 123.5 (t, J=1.4 Hz), 115.4 (t, J=238.3 Hz).
2-(Difluoromethyl)naphthalene (3o):[4] The product (85 mg, 84% yield) was purified with silica gel chromatography (petroleum ether) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.98 (s, 1H), 7.96~7.85 (m, 3H), 7.66~7.50 (m, 3H), 6.81 (t, J=56.4 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -109.9 (d, J=56.4 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 134.3 (t, J=1.4 Hz), 132.5, 131.6 (t, J=22.2 Hz), 128.9, 128.5, 127.9, 127.4, 126.8, 125.9 (t, J=7.5 Hz), 122.0 (t, J=4.8 Hz), 115.0 (t, J=238.5 Hz).
Ethyl 5-(difluoromethyl)nicotinate (3p):[14] The reaction was carried out with ethyl 5-bromonicotinate (2.0 equiv), 1 (0.6 mmol, 1.0 equiv), Ni(PPh3)Br2 (5 mol%), 4, 4'-ditBu- bpy (5 mol%), and molecular sieves (3 Å, 200 mg) in DMPU (3 mL) at 60 ℃ for 12 h. The product (81 mg, 67% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=8:1] as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 9.31 (s, 1H), 8.90 (s, 1H), 8.42 (s, 1H), 6.76 (t, J=55.6 Hz, 1H), 4.43 (q, J=7.1 Hz, 2H), 1.41 (t, J=7.1 Hz, 3H); 19F NMR (376 MHz, CDCl3) δ: -112.9 (d, J=55.6 Hz, 2F); 13C NMR (125.7 MHz, CDCl3) δ: 164.3, 152.8, 150.5 (t, J=6.4 Hz), 134.4 (t, J=5.7 Hz), 130.0 (t, J=23.5 Hz), 126.4, 112.8 (t, J=240.3 Hz), 61.9, 14.2; IR (film) νmax: 3434, 2986, 1728, 1293, 1214, 1027 cm-1; MS (EI) m/z (%): 201 (M+), 156(100); HRMS calcd for C9H9NO2F2 (M+) 201.0601, found 201.0605.
5-(Difluoromethyl)-2-methoxypyridine (3q):[6] The reaction was carried out with 5-bromo-2-methoxypyridine (2.0 equiv.), 1 (0.6 mmol, 1.0 equiv.), Ni(PPh3)Br2 (5 mol%), 4, 4-ditBu-bpy (5 mol%), and molecular sieves (3 Å, 200 mg) in DMPU (3 mL) at 60 ℃ for 12 h. The product (67% yield, determined by 19F NMR) was characterized by 19F NMR and GC-MS analysis. 19F NMR (376 MHz, CDCl3) δ: -109.6 (d, J=56.4 Hz, 2F); GC-MS m/z (%): 159.0 (M+), 158 (100), 140, 129.
2-(Benzyloxy)-5-(difluoromethyl)pyridine (3r):[5] The reaction was carried out with 2-benzyloxy-5-bromopyridine (2.0 equiv.), 1 (0.6 mmol, 1.0 equiv.), Ni(PPh3)Br2 (5 mol%), 4, 4-ditBu-bpy (5 mol%), and molecular sieves (3 Å, 200 mg) in DMPU (3 mL) at 60 ℃ for 12 h. The product (66 mg, 47% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=10:1] as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.30 (d, J=1.6 Hz, 1H), 7.74 (dd, J=8.6 Hz, 2.3 Hz, 1H), 7.46 (d, J=7.1 Hz, 2H), 7.43~7.29 (m, 3H), 6.88 (d, J=8.6 Hz, 1H), 6.65 (t, J=56.0 Hz, 1H), 5.42 (s, 2H); 19F NMR (376 MHz, CDCl3) δ: -109.7 (d, J=56.4 Hz, 2F); 13C NMR (126 MHz, CDCl3) δ: 165.1 (t, J=1.3 Hz), 145.2 (t, J=7.5 Hz), 136.8, 135.9 (t, J=4.5 Hz), 128.5, 128.0, 128.0, 123.6 (t, J=23.4 Hz), 113.8 (t, J=237.6 Hz), 111.7, 68.1; IR (film) νmax: 2956, 1614, 1500, 1349, 1288, 1081, 1017 cm-1; MS (EI) m/z (%): 235 (M+), 91(100); HRMS calcd. for C13H11NOF2 235.0809, found 201.0802.
3-(Difluoromethyl)quinoline (3s):[14] The product (67 mg, 62% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=8:1] as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 9.03 (d, J=1.9 Hz, 1H), 8.29 (s, 1H), 8.16 (d, J=8.5 Hz, 1H), 7.89 (d, J=8.2 Hz, 1H), 7.83~7.78 (m, 1H), 7.65~7.60 (m, 1H), 6.88 (t, J=55.8 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -111.6 (d, J=55.8 Hz, 2F). 13C NMR (125.7 MHz, CDCl3) δ: 149.0 (t, J=1.5 Hz), 147.1 (t, J=5.3 Hz), 133.9 (t, J=6.7 Hz), 131.0, 129.5, 128.3, 127.6, 127.1 (t, J=22.8 Hz), 126.8, 113.7 (t, J=239.5 Hz); IR (film) νmax: 3420, 3061, 2964, 1625, 1500, 1180, 1089, 1032 cm-1; MS (EI) m/z (%): 179 (M+), 179(100); HRMS calcd for C10H7NF2 179.0547, found 179.0541.
6-(Difluoromethyl)-2-methylquinoline (3t):[6] The product (76 mg, 66% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=8:1] as white solid (m.p. 48~50 ℃). 1H NMR (400 MHz, CDCl3) δ: 8.10~8.01 (m, 2H), 7.87 (s, 1H), 7.76 (d, J=8.8 Hz, 1H), 7.31 (d, J=8.4 Hz, 1H), 6.78 (t, J=56.3 Hz, 1H), 2.74 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -110.2 (d, J=56.4 Hz, 2F); 13C NMR (125.7 MHz, CDCl3) δ: 160.7, 148.6 (t, J=1.5 Hz), 136.5, 131.4 (t, J=22.5 Hz), 129.6, 125.7 (t, J=4.9 Hz), 125.7, 125.4 (t, J=7.2 Hz), 122.8, 114.5 (t, J=238.9 Hz), 25.4; IR (film) νmax: 2997, 1631, 1604, 1484, 1086, 1019 cm-1; MS (EI) m/z (%): 193 (M+), 193 (100); HRMS calcd. for C11H9NF2 193.0703, found 193.0695.
6-(Difluoromethyl)isoquinoline (3u): The product (63 mg, 59% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=8:1] as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 9.29 (s, 1H), 8.59 (d, J=5.7 Hz, 1H), 8.03 (d, J=8.5 Hz, 1H), 7.93 (s, 1H), 7.68 (t, J=6.6 Hz, 2H), 6.79 (t, J=56.0 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -111.7 (d, J=56.4 Hz, 2F); 13C NMR (125.7 MHz, CDCl3) δ: 152.4, 143.9, 135.9 (t, J=22.4 Hz), 135.1, 129.1, 128.6, 124.3 (t, J=7.4 Hz), 123.5 (t, J=4.9 Hz), 120.8, 114.1 (t, J=239.8 Hz); IR (film) νmax: 3393, 1591, 1352, 1179, 1034 cm-1; MS (EI) m/z (%): 179 (M+), 179 (100); HRMS calcd. for C10H7NF2 179.0547, found 179.0545.
2-(4-(Difluoromethyl)phenyl)benzo[d]thiazole (3v): The reaction was carried out with 1-(2-benzothiazolyl)-4-bromo- benzene (2.0 equiv.), 1 (0.6 mmol, 1.0 equiv.), Ni(PPh3)Br2 (5 mol%), 4, 4'-ditBu-bpy (5 mol%), and molecular sieves (3 Å, 200 mg) in DMPU (3 mL) at 60 ℃ for 12 h. The product (116 mg, 74% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)= 5:1] as a white solid. m.p. 103~105 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.16 (d, J=8.2 Hz, 2H), 8.09 (d, J=8.1 Hz, 1H), 7.90 (d, J=8.0 Hz, 1H), 7.62 (d, J=8.1 Hz, 2H), 7.57~7.46 (m, 1H), 7.45~7.36 (m, 1H), 6.70 (t, J=56.3 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -111.6 (d, J=56.4 Hz, 2F); 13C NMR (125.7 MHz, CDCl3) δ: 166.6, 154.0, 136.4 (t, J=22.5 Hz), 135.7 (t, J=2.0 Hz), 135.1, 127.7, 126.5, 126.2 (t, J=6.1 Hz), 125.5, 123.4, 121.6, 114.1 (t, J=239.4 Hz); IR (film) νmax: 3054, 1484, 1376, 1073, 1015 cm-1; MS (EI) m/z (%): 261 (M+), 261 (100); HRMS calcd for C14H9NSF2 261.0424, found 261.0422.
2-(4-(Difluoromethyl)phenyl)benzo[d]oxazole (3w):[5] The reaction was carried out with 2-(4-bromophenyl)-1, 3- benzoxazole (2.0 equiv.), 1 (0.6 mmol, 1.0 equiv.), Ni(PPh3)- Br2 (5 mol%), 4, 4'-ditBu-bpy (5 mol%), and molecular sieves (3 Å, 200 mg) in DMPU (3 mL) at 60 ℃ for 12 h. The product (93 mg, 63% yield) was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=5:1] as a white solid. m.p. 98~100 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.33 (d, J=8.1 Hz, 2H), 7.85~7.75 (m, 1H), 7.66 (d, J=8.1 Hz, 2H), 7.62~7.55 (m, 1H), 7.40~7.32 (m, 2H), 6.71 (t, J=56.2 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -111.9 (d, J=56.4 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 161.9 (t, J=1.0 Hz), 150.8, 141.9, 137.0 (t, J=22.5 Hz), 129.4 (t, J=2.0 Hz), 127.8, 126.1 (t, J=6.1 Hz), 125.5, 124.8, 120.2 (d, J=1.0 Hz), 114.1 (t, J=239.6 Hz), 110.7; IR (film) νmax: 3064, 1557, 1454, 1073, 1053, 1015 cm-1; MS (EI) m/z (%): 245 (M+), 245 (100); HRMS calcd for C14H9NOF2 245.0652, found 245.0653.
(13S)-13-Methyl-17-oxo-7, 8, 9, 11, 12, 13, 14, 15, 16, 17-decahydro-6H-cyclopenta[a]phenanthren-3-yl 4-(difluoro- methyl)benzoate (3x): To a 25 mL Schlenk tube were added 2x (0.45 mmol, 1.5 equiv.), Ni(PPh3)2Br2 (5 mol%), 4, 4'-ditBu-bpy (5 mol%), zinc dust (2.0 equiv.), KI (0.4 equiv.) and 3 Å MS (400 mg). The mixture was evacuated and backfilled with argon for three times, then DMPU (1.5 mL) and BrCF2H 1 (2.5 mol•L-1 in 1, 4-dioxane, 0.3 mmol, 1.0 equiv.) were added subsequently. The Schlenk tube was screw capped and put into a preheated oil bath (60 ℃). After stirring overnight, the reaction mixture was cooled to room temperature and filtered with a pad of celite. The filtrate was concentrated, and the residue was purified with silica gel chromatography [V(petroleum ether):V(EtO-Ac)=58:1] to give product 3x (64 mg, 50% yield) as a white solid. m.p. 140~143 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.28 (d, J=8.1 Hz, 2H), 7.66 (d, J=8.1 Hz, 2H), 7.35 (d, J=8.4 Hz, 1H), 6.99 (d, J=8.4 Hz, 1H), 6.96 (s, 1H), 6.73 (t, J=56.1 Hz, 1H), 2.99~2.92 (m, 2H), 2.57~2.47 (m, 1H), 2.47~2.40 (m, 1H), 2.37~2.27 (m, 1H), 2.22~1.94 (m, 4H), 1.71~1.42 (m, 6H), 0.93 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -112.4 (d, J=56.4 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 220.7, 164.6, 148.6, 139.0 (t, J=22.5 Hz), 138.2, 137.7, 131.9, 130.5, 126.5, 125.8 (t, J=6.1 Hz), 121.6, 118.7, 113.9 (t, J=239.9 Hz), 50.5, 47.9, 44.2, 38.0, 35.9, 31.6, 29.4, 26.3, 25.8, 21.6, 13.8; IR (film) νmax: 2934, 2862, 1734, 1492, 1270, 1220, 1067 cm-1; MS (EI) m/z (%): 424 (M+), 155 (100); HRMS calcd for C26H26O3F2 424.1850, found 424.1855.
4-(Difluoromethyl)benzyl 2-(4-isobutylphenyl) propanoate (3y): To a 25 mL Schlenk tube were added 2y (0.45 mmol, 1.5 equiv.), Ni(PPh3)2Br2 (5 mol%), 4, 4'-ditBu- bpy (5 mol%), zinc dust (2.0 equiv.), KI (0.4 equiv.) and 3 Å MS (400 mg). The mixture was evacuated and backfilled with argon for three times, then DMPU (1.5 mL) and BrCF2H 1 (2.5 mol•L-1 in 1, 4-dioxane, 0.3 mmol, 1.0 equiv.) were added subsequently. The Schlenk tube was screw capped and put into a preheated oil bath (60 ℃). After stirring overnight, the reaction mixture was cooled to room temperature and filtered with a pad of celite. The filtrate was concentrated, and the residue was purified with silica gel chromatography [V(petroleum ether):V(EtOAc)=10:1] to give product 3y (64 mg, 62% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.44 (d, J=7.9 Hz, 2H), 7.29 (d, J=8.0 Hz, 2H), 7.21 (d, J=8.0 Hz, 2H), 7.11 (d, J=8.0 Hz, 2H), 6.62 (t, J=56.4 Hz, 1H), 5.15 (s, 2H), 3.78 (q, J=7.1 Hz, 1H), 2.47 (d, J=7.2 Hz, 2H), 1.93~1.81 (m, 1H), 1.53 (d, J=7.2 Hz, 3H), 0.92 (d, J=6.6 Hz, 6H); 19F NMR (376 MHz, CDCl3) δ: -110.7 (d, J=56.4 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 174.3, 140.7, 138.9 (t, J=2.0 Hz), 137.4, 134.0 (t, J=22.4 Hz), 129.3, 127.7, 127.2, 125.7 (t, J=6.1 Hz), 114.5 (t, J=238.7 Hz), 65.5, 45.1, 45.0, 30.2, 22.3, 18.3; IR (film) νmax: 2956, 2869, 1735, 1381, 1158, 1074, 1021 cm-1; MS (EI) m/z (%): 346 (M+), 161 (100); HRMS calcd for C21H24O2F2 346.1744, found 346.1741.
4.4 Gram-scale synthesis of compound 3t
To a 100 mL of Schlenk tube were added 6-bromo-2- methylquinoline (13.5 mmol, 1.5 equiv.), Ni(PPh3)2Br2 (10 mol%), bpy (10 mol%), zinc dust (2.0 equiv.) and KI (0.4 equiv.). The mixture was evacuated and backfilled with argon for three times, DMPU (45 mL) and BrCF2H 1 (2.5 mol•L-1 in 1, 4-dioxane, 9 mmol, 1.0 equiv.) were added subsequently. The Schlenk tube was screw capped and put into a preheated oil bath (60 ℃). After stirring overnight, the reaction mixture was cooled to room temperature. The reaction mixture was filtered with a pad of celite. The filtrate was added into 50 mL of water. The aqueous layer was extracted with a mixture of petroleum ether and EtOAc (V:V=5:1; 100 mL×3). The combined organic layers were dried over anhydrous Na2SO4 and then evaporated under reduced pressure and the resulting crude product was purified by column chromatography with ethyl acetate and petroleum ether (V:V=1:5) as eluent to give product 3t (1.12 g, 64%) as a white solid (m.p. 48~50 ℃).
4.5 Radcial clock experiment
To a 25 mL of Schlenk tube were added 2g (0.9 mmol, 1.5 equiv.), Ni(PPh3)2Br2 (10 mol%), bpy (10 mol%), KI (0.4 equiv.), and Zn dust (2.0 equiv.) under air. The mixture was then evacuated and backfilled with Ar (3 times). Compound 5 (0.9 mmol, 1.5 equiv.), DMPU (3 mL) and BrCF2H (2.5 mol•L-1 in dioxane, 240 μL, 0.6 mmol, 1.0 equiv) were added subsequently. The Schlenk tube was screw capped and put into a preheated oil bath (60 ℃). After stirring for 6 h, the reaction mixture was cooled to room temperature, diluted with ethyl acetate, and filtered through a pad of celite. The filtrate was concentrated, and the residue was washed with 10 mL of water, extracted with organic solvent [V(petroleum ether):V(EtOAc)=20:1] for three times. The organic layer was washed with brine, dried over Na2SO4 and concentrated. The residue was purified by silica gel (petroleum ether) to give compound 6[6] (16 mg, 14% yield). 1H NMR (400 MHz, CDCl3) δ: 7.22~7.11 (m, 4H), 6.02 (t, J=4.4 Hz, 1H), 5.91 (tt, J=56.8, 4.8 Hz, 1H), 2.97 (td, J=16.5, 3.8 Hz, 2H), 2.75 (t, J=8.1 Hz, 2H), 2.28 (dd, J=12.5, 8.0 Hz, 2H); 19F NMR (376 MHz, CDCl3) δ: -114.0 (dt, J=56.8, 16.5 Hz, 2F); MS (EI) m/z (%): 194 (M+), 129 (100); HRMS calcd for C12H12F2 194.0907, found 194.0909.
Supporting Information Optimization of the reaction conditions, mechanistic studies and copies of 1H NMR, 19F NMR and 13C NMR spectra of compounds 3 and 6. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
-
-
[1]
(a) Meanwell, N. A. J. Med. Chem. 2011, 54, 2529.
(b) Erickson, J. A.; McLoughlin, J. I. J. Org. Chem. 1995, 60, 1626. -
[2]
(a) Ge, S.; Chaladaj, W.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 4149.
(b) Gu, Y.; Leng, X.; Shen, Q. Nat. Commun. 2014, 5, 5405.
(c) Xu, L.; Vicic, D. A. J. Am. Chem. Soc. 2016, 138, 2536.
(d) Serizawa, H.; Ishii, K.; Aikawa, K.; Mikami, K. Org. Lett. 2016, 18, 3686. -
[3]
(a) Moore, G. G. I. J. Org. Chem. 1979, 44, 1708.
(b) Ruppert, I.; Schlich, K.; Volbach, W. Tetrahedron Lett. 1984, 25, 2195.
(c) Tyutyunov, A. A.; Boyko, V. E.; Igoumnov, S. M. Fluorine Notes 2011, 74, 1.
(d) Prakash, G. K. S.; Hu, J.; Olah, G. A. J. Org. Chem. 2003, 68, 4457. -
[4]
Feng, Z.; Min, Q.-Q.; Zhang, X. Org. Lett. 2016, 18, 44. doi: 10.1021/acs.orglett.5b03206
-
[5]
Feng, Z.; Min, Q.-Q.; Fu, X.-P.; An, L.; Zhang, X. Nat. Chem. 2017, 9, 918. doi: 10.1038/nchem.2746
-
[6]
Xu, C.; Guo, W.-H.; He, X.; Guo, Y.-L.; Zhang, X.-Y.; Zhang, X. Nat. Commun. 2018, 9, 1170. doi: 10.1038/s41467-018-03532-1
-
[7]
Guiadeen, D.; Kothandaraman, S.; Yang, L.; Mills, S. G.; MacCoss, M. Terahedron Lett. 2008, 49, 6368. doi: 10.1016/j.tetlet.2008.08.083
-
[8]
Fu, X.-P.; Xiao, Y.-L.; Zhang, X. Chin. J. Chem. 2018, 36, 143. doi: 10.1002/cjoc.201700624
-
[9]
Sheng, J.; Ni, H.-Q.; Bian, K.-J.; Li, Y.; Wang, Y.-N.; Wang, X.-S. Org. Chem. Front. 2018, 5, 606. doi: 10.1039/C7QO00934H
-
[10]
(a) Feng, Z.; Xiao, Y.-L.; Zhang, X. Acc. Chem. Res. 2018, 51, 2264.
(b) Feng, Z.; Chen, F.; Zhang, X. Org. Lett. 2012, 14, 1938.
(c) Feng, Z.; Min, Q.-Q.; Xiao, Y.-L.; Zhang, B.; Zhang, X. Angew. Chem., Int. Ed. 2014, 53, 1669.
(d) Min, Q.-Q.; Yin, Z.; Feng, Z.; Guo, W.-H.; Zhang, X. J. Am. Chem. Soc. 2014, 136, 1230.
(e) Xiao, Y.-L.; Guo, W.-H.; He, G.-Z.; Pan, Q.; Zhang, X. Angew. Chem., Int. Ed. 2014, 53, 9909. -
[11]
Prinsell, M. R.; Everson, D. A.; Weix, D. J. Chem. Commun. 2010, 46, 5743.
-
[12]
19F NMR showed the chemical shifts of difluoromethyl Zinc species A1 and A2 are consistent with the literature, see: Burton, D. J.; Hartgraves, G. A. J. Fluorine Chem. 2007, 128, 1198.
-
[13]
(a) Weix, D. Acc. Chem. Res. 2015, 48, 1767.
(b) Gu, J.; Wang, X.; Xue, W.; Gong, H. Org. Chem. Front. 2015, 3, 1411. -
[14]
Lu, C.-H, ; Gu, Y.; Wu, J.; Gu, Y.-C.; Shen, Q.-L. Chem. Sci. 2017, 8, 4848. doi: 10.1039/C7SC00691H
-
[1]
-
Table 1. Representative results for optimization of Ni-catalyzed difluoromethylation of 2a with BrCF2H (1)a
Entry [Ni] Zn/equiv. Additive (equiv.) Yieldb/% of 3a/4/1 1 NiBr2•Diglyme 1.2 — 16/10/36 2 NiBr2•Diglyme 1.2 LiI (0.2) 24/Trace/44 3 NiBr2•Diglyme 1.2 NaI (0.2) 30/12/21 4 NiBr2•Diglyme 1.2 KI (0.2) 45/Trace/25 5 NiBr2•Diglyme 2.0 KI (0.2) 62/Trace/7 6 NiBr2•Diglyme 2.0 KI (0.4) 66/5/0 7 Ni(PPh3)2Br2 2.0 KI (0.4) 73/5/0 8 Ni(PPh3)2Cl2 2.0 KI (0.4) 60/12/0 9 Ni(dppe)Cl2 2.0 KI (0.4) 27/12/0 10 NiCl2•DME 2.0 KI (0.4) 19/Trace/34 11 Ni(cod)2 2.0 KI (0.4) 0/8/45 12c Ni(PPh3)2Br2 2.0 KI (0.4) 78/4/0 13c, d Ni(PPh3)2Br2 2.0 KI (0.4) 90 (91)/Trace/0 14c, d — 2.0 KI (0.4) 0/9/0 15c, d, e Ni(PPh3)2Br2 2.0 KI (0.4) 0/8/10 a Reaction conditions (unless otherwise specified): 1 (0.3 mmol, 1.0 equiv.), 2a (0.75 mmol, 2.5 equiv.), DMPU (2 mL), 6 h. b Determined by 19F NMR using fluorobenzene as an internal standard and number in parenthesis is isolated yield. c Ni(PPh3)2Br2 (10 mol%), bpy (10 mol%), 60 ℃, 6 h. d 1 (0.3 mmol, 1.0 equiv.), 2a (0.45 mmol, 1.5 equiv.), DMPU (1.5 mL). e The reaction was run in the absence of bpy.  下载: 导出CSV
下载: 导出CSV
Table 2. Ni-catalyzed reductive cross-coupling of bromodifluoromethane (1) with aryl bromides (2)a
aReaction conditions (unless otherwise specified): 2 (0.9 mmol, 1.5 equiv.), 1 (0.6 mmol, 1.0 equiv.), DMPU (3.0 mL), 60 ℃, 6 h. Yields of isolated products are given. bNMR yield determined by 19F NMR. c2 (2.0 equiv.), 1 (0.6 mmol, 1.0 equiv.), Ni(PPh3)Br2 (5 mol%), 4, 4'-ditBu-bpy (5 mol%), 3Å MS (200 mg), 12 h. dGram-scale synthesis and reaction run for 12 h. e2 (1.5 equiv.), 6 (0.3 mmol, 1.0 equiv.), Ni(PPh3)Br2 (5 mol%), 4, 4'-ditBu-bpy (5 mol%), 3Å MS (400 mg), 12 h.
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 19
- 文章访问数: 2079
- HTML全文浏览量: 211
