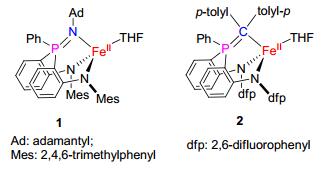
图 1.
磷亚胺和磷叶立德配位的亚铁配合物
Figure 1.
Iminophosphorane-iron(Ⅱ) and phosphonium ylide- iron(Ⅱ) complexes

第15族元素磷、砷、锑、铋的亚胺化合物R3MNR' (M=P, As, Sb, Bi; R, R'=烷基、芳基)是叶立德化合物R3M=CR'2和氧化物R3M=O的类似物[1].磷亚胺化合物比较稳定, 其合成和反应化学研究较多[1], 并且磷亚胺在配位化学中是应用较为广泛的含氮配体[2].砷、锑、铋的亚胺化合物反应活性较高, 在已报道研究中它们常作为活性中间体, 通过原位生成的方式与底物发生乃春转移反应[3~6].与磷亚胺相比, 砷、锑、铋亚胺化合物的M—N键高度极化, 其有效稳定通常需要通过氮上引入吸电子取代基来实现[3~6].文献中分子结构得到表征的砷、锑、铋的亚胺化合物相对较少, 且多为亚胺氮上取代基为吸电性的酰基、磺酰基, 如Roesky等[3]报道的磺酰亚胺化合物Ph3AsNSO2N(S3N2)、Matano等[4]报道的三氯乙酰亚胺Ar3MNCOCCl3和三氟甲磺酰亚胺化合物Ph3MNSO2CF3 (M=As, Sb, Bi)[5].然而, 砷、锑、铋的亚胺化合物作为配体与金属形成配合物未见报道.
在前期的研究中, 我们发现二胺基-膦配体支撑的亚铁配合物可与有机叠氮化合物和有机重氮化合物反应生成相应的磷亚胺和磷叶立德配位的亚铁配合物1和2(图 1), 其中磷叶立德配位的亚铁配合物可以转化为开壳型铁卡宾中间体与烯烃作用生成环丙烷化产物[7, 8].考虑到砷亚胺金属配合物未见报道, 我们设想通过二胺基-胂配体支撑的亚铁配合物与有机叠氮化合物的反应来合成相应的砷亚胺亚铁配合物.本文将介绍我们利用该方法实现的砷亚胺亚铁配合物的合成和结构表征, 并对其乃春转移反应进行的初步探索.
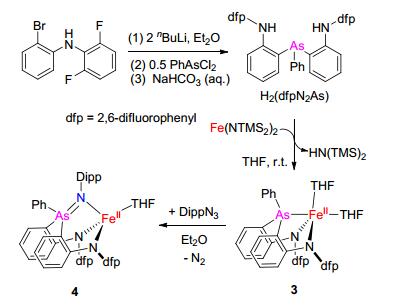
依据以前报道的磷桥联二苯胺配体H2(dfpN2P)的合成方法[8], 由(2, 6-F2C6H3)(2-BrC6H4)NH与2 equiv.正丁基锂反应原位形成二锂化合物(2, 6-F2C6H3)(2-LiC6H4)- NLi后与0.5 equiv.的PhAsCl2反应, 以46%的产率合成得到了砷桥联的二苯胺配体((o-(N-(2, 6-F2C6H3)NH)- C6H4)2AsPh即H2(dfpN2As) (Scheme 1), 并对其进行了核磁共振氢谱、碳谱、氟谱、红外光谱和ESI-质谱表征.砷桥联的二苯胺配体H2(dfpN2As)与0.5 equiv. [Fe(N-(TMS)2)2]2在四氢呋喃(THF)中反应生成二胺基-胂配位的亚铁配合物[(κ-N, N, As-dfpN2As)Fe(THF)2] (3), 分离产率为38% (Scheme 1).核磁反应跟踪显示H2(dfpN2As)与[Fe(N(TMS)2)2]2反应可以几乎定量生成配合物3.配合物3较低的分离产率主要是由于它在常见的低极性溶剂如甲苯、乙醚、四氢呋喃中有较好的溶解性.
在乙醚中缓慢挥发重结晶, 可得配合物3的橙色晶体.单晶X射线衍射确定其结构与[(κ-N, N, P-dfpN2P)-Fe(THF)2][8]相似, 为五配位的亚铁配合物(CCDC: 1827395).如图 2所示, 配合物3中二胺基-胂配体以fac-形式与铁中心配位, Fe—N和Fe—As键键长分别为2.038(7), 2.038(7)和0.2562(3) nm, As—Fe—N和N— Fe—N键角分别为81.3(2)°和121.6(4)°.与配合物3的高自旋的电子结构特征相关, 其Fe—As键键长比低自旋铁有机砷(Ⅲ)配合物中相应键长长, 如Fe(CO)4- (As(o-tolyl)3) [0.2393(1) nm][9], (OC)3Fe(µ-CHCHPh)- (µ-SBut)Fe(CO)2(AsPh3) [0.2368(1) nm][10], CpFe(CO)- (AsPh3)SCO(2-C4H3S) (0.233 nm)[11], [Fe(NO)(o-C6H4-(AsMe2)2)][ClO4]2 [0.2381(3)~0.2416(3) nm[12].配合物3的57Fe Mössbauer谱值(δ=0.88 mm/s, |ΔEQ|=1.50 mm/s at 80 K)与溶液相磁矩测试结果(C6D6溶液中μeff=4.4(1) μB)也表明其为高自旋态(S=2)二价铁配合物.
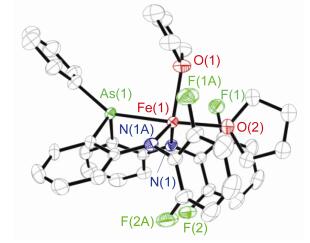
Selected distances (nm) and angles (°): Fe(1)—N(1) 0.2038(7), Fe(1)—N(1A) 0.2038(7), Fe(1)—O(1) 0.2227(10), Fe(1)—O(2) 0.2168(10), Fe(1)—As(1) 0.2562(3), N(1)—Fe(1)—N(1A) 121.6(4), N(1)—Fe(1)—O(1) 117.9(2), N(1)—Fe(1)—O(2) 100.1(2), N(1)—Fe(1)—As(1) 81.3(2), As(1)—Fe(1)—O(1) 91.5(3), As(1)—Fe(1)—O(2) 177.0(3), O(1)—Fe(1)—O(2) 85.4(4)
配合物3可与有机叠氮反应生成胂亚胺配位的亚铁配合物.室温下向配合物3的乙醚溶液中滴加DippN3的乙醚溶液时, 溶液逐渐由橙色变为绿色, 同时有气泡冒出.反应结束后通过重结晶可得棕绿色晶状固体.单晶X射线衍射确定其为砷亚胺配位的亚铁配合物[(κ-N, N, N-dfpN2AsNDipp)Fe(THF)2] (4) (Scheme 1).配合物4为首例砷亚胺金属配合物(CCDC: 1827396).它为顺磁性配合物, 其57Fe Mössbauer谱参数[δ=0.90 mm/s, |ΔEQ|=2.53 mm/s (80 K)]和溶液相磁矩[5.2(1) μB in C6D6]共同表明它具有高自旋态(S=2)二价铁电子结构特征.配合物4的形成可能是由(二胺基-胂)亚铁配合物3先与DippN3反应生成(二胺基-胂)铁乃春配合物中间体(κ-N, N, As-dfpN2As)Fe(NDipp), 后者进一步发生分子内的乃春转移反应导致.对照反应显示配体前体H2(dfpN2As)与DippN3在室温或80 ℃下均不能反应.这说明3中胂配体部分直接与叠氮反应生成砷亚胺再与铁中心配位形成4的可能性较小.
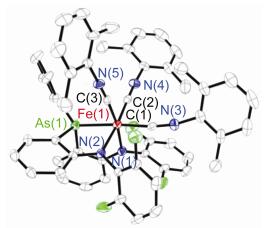
配合物4的分子结构如图 3所示.其三齿型二胺基-胂亚胺配体的三个氮原子以fac-形式与铁中心配位. Fe—N(亚胺)键键长为0.2008(3) nm, Fe—N(胺基)键键长为0.2015(4)和0.1996(3) nm.这些Fe—N键键长与磷亚胺亚铁配合物[(κ-N, N, N-MesN2PNAd)Fe(THF)] (1)中相应键长相当[7].配合物4中Fe—As间距离为0.285 nm, 明显长于3中Fe—As键长[0.2562(3) nm], 也比砷与高自旋铁的共价半径之和[0.271 nm]略长. As—N(亚胺)键键长为0.1752(3) nm, 与已报道的一些砷亚胺配合物和砷铵盐中As—N键键长相当, 如Ph3AsNSO2N(S3N2) [0.1760(2) nm][3], Ph3AsNCOCCl3 [0.1783(2) nm][4], Ph3As-NSO2CF3 [0.1776(7) nm][5]和[Ph3AsNAsPh3][Cl] [0.1749(3) nm][13], 短于典型的As—N单键键长[0.184~1.88 nm][5]. As—N(1)—Fe键角为98.45(2)°, 与[(MesN2PNAd)Fe(THF)]中P—N(1)—Fe键角[100.72(6)°]相当[7].需要指出的是, 尽管单晶衍射数据显示配合物4中As—N键较短, 但这并不能说明其为As=N双键. Koketsu等[14]的理论计算研究表明在H3MNH (M=P, As, Sb, Bi)中离子形式(M+-N-)是主导的共振结构. Lammertsma等[15]对H3PNH的理论研究也指出H3PNH中较短的P—N键主要是由两基团间的库伦相互作用导致, 而其中的π-键贡献较小.基于这些认识, 同时考虑到亚铁中心的配位形成Fe2+…-N(Dipp)—+AsAr3可稳定极化的砷亚胺, 我们认为4中的As—N键也可能主要是单键性质.
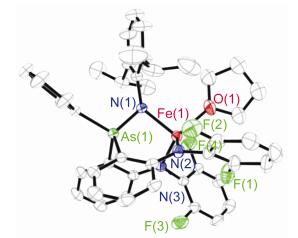
Selected distances (nm) and angles (°): Fe(1)—N(1) 0.2008(3), Fe(1)—N(2) 0.2015(4), Fe(1)—N(3) 0.1996(3), As(1)—N(1) 0.1752(3), Fe(1)—O(1) 0.2112(4), N(1)—Fe(1)—N(2) 101.60(15), N(1)—Fe(1)—N(3) 99.58(14), N(2)—Fe(1)—N(3) 112.75(16), As(1)—N(1)—Fe(1) 98.45(16), N(1)—Fe(1)—O(1) 100.06(16)
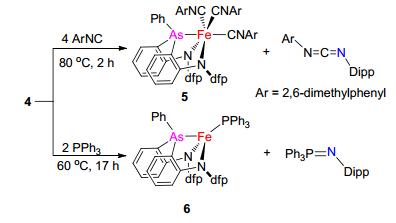
考虑到砷亚胺是比磷亚胺更活泼的乃春转移试剂[4, 5], 我们进一步考察了4与一些有机底物的反应, 发现其可与异腈和膦发生乃春转移反应.配合物4与4 equiv. 2, 6-二甲基苯基异腈于80 ℃下反应生成二胺基-胂配位的亚铁配合物[(κ-N, N, As-dfpN2As)Fe(CNC6H3Me2-2, 6)3] (5)和乃春转移产物碳二亚胺DippNCNC6H3Me2- 2, 6, 二者产率分别为34%和49% (Scheme 2).配合物4与2 equiv. Ph3P于60 ℃下反应生成二胺基-胂配位的亚铁配合物(κ-N, N, As-dfpN2As)Fe(PPh3) (6)和乃春转移产物Ph3PNDipp (Scheme 2).配合物5为红色固体.如图4所示, 单晶X射线衍射表征确认其结构为六配位的亚铁配合物(CCDC: 1827397).其中铁中心与三齿二胺基-胂配体配位外, 还与三分子异腈作用, 呈八面体构型.其Fe—N键键长与3中相应键长相当, Fe—As键[0.2293(1) nm]则比3中Fe—As键短约0.027 nm.三个Fe—C(异腈)键键长分别为0.1811(4), 0.1863(5)和0.1818(4) nm, 与已报道的低自旋亚铁异腈配合物中相应键长相当[16, 17].这些结构数据表明配合物5的基态电子构型可能为低自旋态, 其57Fe Mössbauer谱的同质异能位移(δ=0.05 mm/s)和四极距裂分值(|ΔEQ|=0.40 mm/s)在低自旋亚铁配合物的范围内, 同时配合物5的核磁共振氢谱、碳谱、氟谱均展示出抗磁性特征.配合物6为顺磁性.其溶液相磁矩[μeff=4.8(1) μB]和57Fe Mössbauer谱值(δ=0.65 mm/s, |ΔEQ|=2.23 mm/s)表明其S=2的高自旋电子结构特征.配合物6的组成通过元素分析(C, H, N)得到确认.有机产物碳二亚胺和磷亚胺通过核磁共振表征, 其数据与文献报道一致[18, 19].
Selected distances (nm) and angles (°): Fe(1)—N(1) 0.2052(3), Fe(1)—N(2) 0.2070(3), As(1)—Fe(1) 0.22927(6), Fe(1)—C(1) 0.1864(4), Fe(1)—C(2), 0.1811(4), Fe(1)—C(3) 0.1818(4), N(1)—Fe(1)—N(2) 91.5(1), N(1)—Fe(1)—C(1) 95.3(2), N(1)—Fe(1)—C(2) 90.7(1), N(1)—Fe(1)—C(3) 171.7(1), N(1)—Fe(1)—As(1) 83.02(9), N(2)—Fe(1)—C(1) 96.5(1), N(2)—Fe(1)—C(2) 174.5(1), N(2)—Fe(1)—C(3) 85.2(1), N(2)—Fe(1)—As(1) 79.19(9), As(1)—Fe(1)—C(1) 175.3(1), As(1)—Fe(1)—C(2) 96.1(1), As(1)—Fe(1)—C(3) 88.9(1)
值得一提的是, 与配合物4结构相似的膦亚胺亚铁配合物[(κ-N, N, N-MesN2PNAd)Fe(THF)][7]在相同的条件下与CNC6H3Me2-2, 6和PPh3均不发生乃春转移反应.这说明砷亚胺亚铁配合物具有更高的活性, 与未配位的砷亚胺的反应活性较磷亚胺高的认知一致[4, 5, 14].除与异腈和膦的反应外, 我们也考察了4与1-辛烯、苯乙烯、3-己炔在室温或80 ℃下的反应, 但均未观察到乃春转移产物的生成.考虑人们对砷亚胺配合物的认知极其有限, 我们也在试图利用二胺基-胂配合物与有机叠氮化合物的反应合成其它砷亚胺过渡金属配合物.
通过邻胺基取代苯基锂与PhAsCl2的反应合成了二胺基-胂配体H2(dfpN2As).通过H2(dfpN2As)与[Fe(N-(TMS)2)2]2反应合成了二胺基-胂配位的亚铁配合物[(κ-N, N, As-dfpN2As)Fe(THF)2], 进一步通过二胺基-胂配位的亚铁配合物与有机叠氮DippN3反应合成了二胺基-砷亚胺配位的亚铁配合物[(κ-N, N, N-dfpN2AsNDipp)-Fe(THF)].这些配合物均通过系列谱学方法进行了表征.其中的砷亚胺亚铁配合物是首例砷亚胺过渡金属配合物.反应化学研究显示该砷亚胺亚铁配合物可以与2, 6-二甲基苯基异腈和三苯基膦发生乃春转移反应生成二胺基-胂配位的亚铁配合物[(κ-N, N, As-dfpN2As)Fe-(CNC6H3Me2-2, 6)3]和[(κ-N, N, As-dfpN2As)Fe(PPh3)]以及相应的有机产物DippNCNC6H3Me2-2, 6与DippNPPh3.这些从(二胺基-胂)亚铁配合物到(二胺基-砷亚胺)亚铁配合物的转化的实现显示了砷配体在发展新型铁催化基团转移反应中具有的潜在应用价值.
1H NMR, 13C NMR, 19F NMR和31P NMR在VARIAN Mercury 400 MHz或Agilent 400 MHz核磁共振仪上测定. 1H NMR和13C NMR以氘代试剂的残余峰定标, 19F NMR的化学位移以CF3Cl3(外标)为零点定标.元素分析、高分辨质谱由中国科学院上海有机化学研究所元素分析组测定及质谱中心测定.低温单晶X射线衍射在Bruker APEX CCD上测定, 具体方法:于手套箱中挑选合适的单晶用Paratone-N型油脂包裹, 将单晶放置在玻璃纤维顶端并置于低温氮气气流中, 在石墨单色Mo Kα射线照射下, 用Bruker APEX CCD衍射仪收集晶体的衍射数据.配合物的红外光谱在Nicolet 380 FT-IR上测定:在手套箱中将配合物与适量的KBr混合并研磨均匀, 取出后在空气中快速压片测试.有机物的红外光谱在Bruker TENSOR 27 ART-FTIR上测定.溶液相磁矩在室温下通过Evans方法测定[23, 24].紫外-可见-近红外吸收光谱在Shimadzu UV-3600光谱仪上测定:以溶剂四氢呋喃作为空白, 测试溶液放置于聚四氟乙烯塞密闭的2 mm石英比色皿中, 波长范围为200~2000 nm. 57Fe Mössbauer谱在WSS-10穆斯堡尔光谱仪上测定, 对测试得到的数据, 使用Mosswinn 4.0Pre.软件进行拟合.具体方法是:在手套箱中将待测样品研细后, 以Paratone-N型油脂拌样, 均匀填充在聚四氟乙烯样品池中, 经液氮冷冻固定后, 安置于光谱仪上进行测试.所有与配合物相关实验均在氮气氛围下, 采用标准Schlenk操作或在有氮气氛围的Vigor手套箱中进行.所用溶剂在使用前都通过有机溶剂纯化系统(Innovative Technology)除水后在真空线上氮气鼓泡除去空气, 再经活化后的分子筛进一步干燥, 存储在手套箱中使用. C6D6经Na-K合金干燥后真空转移, 储存在手套箱里使用. (2, 6-F2C6H3)(2-BrC6H4)NH[8], PhAsCl2[20], [Fe(N(TMS)2)2]2[21]和2-azido-1, 3-diisopropylbenzene- (DippN3)[22]根据文献方法制备.
-78 ℃下向(2, 6-F2C6H3)(2-BrC6H4)NH (2.85 g, 10.0 mmol)的乙醚溶液(30 mL)中加入正丁基锂的正己烷溶液(2.5 mol/L, 8.0 mL, 20 mmol).反应体系缓慢恢复至室温, 继续搅拌过夜.反应液冷却至-78 ℃后加入PhAsCl2 (1.10 g, 5.0 mmol), 再将反应混合物缓慢恢复至室温, 继续搅拌48 h, 然后加入饱和的碳酸氢钠水溶液(30 mL)淬灭反应.分出有机相, 水相用二氯甲烷萃取(50 mL×3).合并有机相, 用饱和食盐水洗涤, 无水硫酸钠干燥, 真空除去溶剂, 硅胶柱层析[SiO2, 300~400目, 淋洗剂: V(正己烷):V(二氯甲烷)=8:1]得到1.28 g白色固体粉末, 即为(o-(N-(2, 6-F2C6H3)NH)- C6H4)2AsPh (H2(dfpN2As), 产率46%. 1H NMR (400 MHz, CDCl3, 298 K) δ: 7.54~7.47 (m, 2H), 7.44~7.39 (m, 3H), 7.31~7.24 (m, 2H), 7.13~7.08 (m, 2H), 6.98~6.84 (m, 8H), 6.78 (d, J=8.0 Hz, 2H), 5.68 (s, 2H, NH); 13C NMR (100 MHz, CDCl3, 298 K) δ: 156.33 (dd, JC-F=247.6, 5.6 Hz), 146.64, 135.82, 134.31, 134.23, 130.08, 129.29, 129.22, 125.92, 122.78 (t, JC-F=9.5 Hz), 122.34, 119.87 (t, JC-F=14.8 Hz), 116.19, 111.89 (dd, JC-F=17.6, 5.9 Hz); 19F NMR (376 MHz, CDCl3, 298 K) δ: -121.02; IR (film) ν: 3370 (w) 3351 (w), 2957 (w), 2916 (w), 2851 (w), 1582 (m), 1496 (s), 1467 (s), 1443 (m), 1300 (m), 1237 (m), 1059 (w), 1000 (s), 877 (w), 774 (s), 755 (s), 737 (m), 696 (s) cm-1; HRMS (ESI) calcd for C30H21AsF4N2 (M+H)+ 560.0857, found 560.0847.
室温下向H2(dfpN2As) (237 mg, 0.42 mmol)的四氢呋喃(20 mL)溶液中加入[Fe(N(TMS)2)2]2 (158 mg, 0.21 mmol).反应过夜后得到黄色溶液, 减压除去溶剂, 用正己烷(15 mL)洗涤, 得黄色粉末状固体.该黄色固体在乙醚溶液中挥发重结晶可得122 mg橙黄色晶状固体, 即为[(κ-N, N, As-dfpN2As)Fe(THF)2] (3), 产率38%. UV-vis-NIR (THF) λmax: 278, 1074 nm; 1H NMR (400 MHz, C6D6, 298 K) δ: 46.63 (very br), 37.43 (v1/2=25 Hz), 33.18 (very br), 31.96 (v1/2=91 Hz), 23.82 (v1/2=47 Hz), 16.28 (v1/2=99 Hz), 14.61 (very br), 10.14 (v1/2=351 Hz), 1.47 (v1/2=38 Hz), 0.31 (v1/2=11 Hz), -8.02 (very br), -21.94 (v1/2=26 Hz), -38.79 (v1/2=61 Hz). Anal. calcd for C38H35F4FeN2O2As: C 60.18, H 4.65, N 3.69; found C 59.43, H 4.69, N 3.73. Magnetic susceptibility (C6D6, 298 K): μeff=4.4(1)μB. 57Fe Mössbauer spectrum (Zero-field, 80 K): δ=0.88 mm/s, |ΔEQ|=1.50 mm/s.
室温下向配合物[(κ-N, N, As-dfpN2As)Fe(THF)2] (3, 301 mg, 0.39 mmol)的乙醚(25 mL)溶液中逐滴加入DippN3 (87 mg, 0.42 mmol), 溶液迅速变为棕色, 同时有气泡产生.反应4 h后, 减压除去溶剂, 用正己烷(15 mL)洗涤棕色残余物可得棕绿色粉末状固体.该固体溶于乙醚中, 经挥发重结晶可得棕绿色晶体137 mg, 即为[(κ-N, N, N-dfpN2AsNDipp)Fe(THF)] (4), 产率40%. UV-vis- NIR (THF) λmax: 279, 492, 1255 nm; 1H NMR (400 MHz, C6D6, 294 K) δ: 41.78 (v1/2=35 Hz), 40.14 (very br), 35.45 (v1/2=70 Hz), 29.76 (v1/2=36 Hz), 26.69 (v1/2=76 Hz), 19.06 (very br), 11.54 (v1/2=160 Hz), 9.59 (v1/2=48 Hz), 5.56 (v1/2=65 Hz), 3.94 (very br), 3.58 (v1/2=30 Hz), -2.26 (v1/2=178 Hz), -3.83 (v1/2=60 Hz), -12.76 (v1/2=93 Hz), -27.58 (v1/2=30 Hz), -36.60 (v1/2=44 Hz); IR (KBr) ν: 3051 (w) 2961 (m), 2865 (w), 1581 (m), 1544 (w), 1470 (s), 1435 (s), 1381 (w), 1360 (w), 1316 (m), 1286 (m), 1263 (m), 1191 (m), 1164 (w), 1099 (w), 1079 (w), 1054 (w), 1026 (w), 1002 (s), 941 (w), 895 (w), 865 (w), 778 (m), 740 (m), 716 (m), 692 (w) cm-1. Anal. calcd for C46H44F4FeN3OAs: C 64.12, H 5.15, N 4.88; found C 64.01, H 5.07, N 5.03. Magnetic susceptibility (C6D6, 298 K): μeff=5.2(1)μB. 57Fe Mössbauer spectrum (Zero-field, 80 K): δ=0.90 mm/s, |ΔEQ|=2.53 mm/s.
室温下, 向配合物4 (286 mg, 0.33 mmol)的甲苯(20 mL)溶液中加入2, 6-二甲基苯基异腈(182 mg, 1.38 mmol), 溶液变为棕红色. 80 ℃下反应3 h, 得到棕红色溶液, 减压除去溶剂, 用正己烷(15 mL)洗涤过滤后, 滤液经真空除去溶剂, 硅胶柱层析分离(SiO2, 300~400目, 淋洗剂:正己烷), 得到50 mg无色液体, 即为DippNCNC6H3Me2-2, 6, 产率49%.过滤后所得的546 mg红色粉末状固体, 即为[(κ-N, N, As-dfpN2As)Fe(CN-C6H3Me2-2, 6)3] (5), 产率84%.配合物5的单晶可通向其四氢呋喃溶液中缓慢扩散乙醚得到.
DippNCNC6H3Me2-2, 6: 1H NMR (400 MHz, CDCl3) δ: 7.14 (s, 3H), 7.05 (d, J=7.4 Hz, 2H), 7.00~6.93 (m, 1H), 3.46 (m, 2H), 2.41 (s, 6H), 1.28 (d, J=6.8 Hz, 12H); 13C NMR (100 MHz, CDCl3) δ: 142.92, 136.20, 133.20, 132.63, 128.28, 127.17, 125.18, 124.34, 123.42, 29.21, 23.42, 19.06.该无色液体的1H NMR和13C NMR数据与文献报道一致[18].
[(κ-N, N, As-dfpN2As)Fe(CNC6H3Me2-2, 6)3] (5): UV-vis (THF) λmax: 277, 365, 486 nm; 1H NMR (400 MHz, THF-d8, 298 K) δ: 7.79 (d, J=7.1 Hz, 2H), 7.42 (t, J=10.3 Hz, 1H), 7.27 (d, J=7.4 Hz, 4H), 7.11~7.05 (m, 1H), 7.01~6.95 (m, 4H), 6.93 (d, J=7.4 Hz, 4H), 6.72 (t, J=6.9 Hz, 6H), 6.41 (d, J=7.9 Hz, 2H), 6.19 (t, J=7.1 Hz, 2H), 6.07 (d, J=8.5 Hz, 2H), 2.07 (s, 6H), 1.87 (s, 12H); 13C NMR (100 MHz, THF-d8, 293 K) δ: 177.71, 177.03, 164.15, 162.65 (ddd, 1JC-F=257.2 Hz, 3JC-F=7.3 Hz), 161.84 (dd, 1JC-F=244.5 Hz, 4JC-F=2.4 Hz), 136.62~136.24 (m), 135.92, 135.08, 133.96, 132.59, 131.68, 130.65, 130.51, 129.70, 129.56, 128.38, 128.31, 128.21, 127.71, 121.97~121.78 (m), 121.46, 116.16~115.84 (m), 112.48~112.15 (m), 111.83~111.43 (m), 18.90, 18.28; 19F NMR (376 MHz, THF-d8, 298 K) δ: -112.71, -115.00; IR (KBr) ν: 2140 (s), 2090 (s) cm-1. Anal. calcd for C57H46F4FeN5As: C 67.83, H 4.60, N 6.95; found C 67.82, H 4.75, N 6.72. 57Fe Mössbauer spectrum (Zero-field, 80 K): δ=0.05 mm/s, |ΔEQ|=0.40 mm/s.
室温下向J-Young核磁管中加入配合物4 (13 mg, 0.015 mmol), C6D6 (0.35 mL)和PPh3 (8 mg, 0.030 mmol).室温下反应2 h后1H NMR和31P NMR跟踪显示配合物4的信号峰仍然存在, 且没有新的信号峰产生.在60 ℃下加热1 h后, 1H NMR和31P NMR显示有新物质生成.加热反应17 h后, 配合物4的核磁信号峰消失, 31P NMR谱在δ -9.39处出现Ph3PNDipp的特征信号峰[19]. 1H NMR显示除Ph3PNDipp外有新的顺磁性配合物生成.通过加入均三甲氧基苯(2.4 mg, 0.014 mmol)为内标, 由1H NMR定量确定Ph3PNDipp产率为74% (0.011 mmol).新的顺磁性配合物其氢谱与配合物3与PPh3直接反应生成(κ-N, N, As-dfpN2As)Fe(PPh3) (6)一致.
向配合物3 (313 mg, 0.41 mmol)的甲苯(20 mL)溶液中加入PPh3 (119 mg, 0.45 mmol).室温下反应3 h后, 减压除去溶剂可得红色固体粉末.该固体经正己烷(10 mL)和乙醚(3 mL)洗涤后所得的棕红色固体粉末溶解于四氢呋喃后, 通过乙醚扩散重结晶, 可得(κ-N, N, As- dfpN2As)Fe(PPh3) (6)的红色晶状固体, 197 mg, 产率55%. UV-vis (THF) λmax: 1029 nm; 1H NMR (400 MHz, C6D6, 294 K) δ: 47.38 (v1/2=170 Hz), 43.26 (v1/2=270 Hz), 37.09 (v1/2=227 Hz), 35.01 (v1/2=61 Hz), 30.61 (v1/2=109 Hz), 12.23 (v1/2=117 Hz), 8.91 (very br), -2.35 (v1/2=63 Hz), -21.15 (very br), -22.23 (v1/2=58 Hz), -49.10 (v1/2=138 Hz), -50.51 (v1/2=141 Hz). Anal. calcd for C48H34F4FeN2PAs: C 65.77, H 3.91, N 3.20; found C 65.50, H 4.00, N 3.20. Magnetic susceptibility (C6D6, 294 K): μeff=4.8(1) μB. 57Fe Mössbauer spectrum (Zero-field, 80 K): δ=0.65 mm/s, |ΔEQ|=2.23 mm/s.
辅助材料(Supporting Information) 配合物3~5的晶体结构参数和配合物3~6的57Fe Mössbauer谱参数、核磁共振及紫外可见光谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Patai, S. The Chemistry of Organic Arsenic, Antimony and Bismuth Compounds, Wiley, Chichester, U. K., 1994.
García-Álvarez, J.; García-Garrido, S. E.; Cadierno, V. J. Organomet. Chem. 2014, 751, 792. doi: 10.1016/j.jorganchem.2013.07.009
Roesky, H. W.; Witt, M.; Clegg, W.; Isenberg, W.; Noltemeyer, M.; Sheldric, G. M. Angew. Chem., Int. Ed. Engl. 1980, 19, 943.
Matano, Y.; Nomura, H.; Suzuki, H.; Shiro, M.; Nakano, H. J. Am. Chem. Soc. 2001, 123, 10954. doi: 10.1021/ja003623l
Matano, Y.; Nomura, H.; Suzuki, H. Inorg. Chem. 2002, 41, 1940. doi: 10.1021/ic0110575
Nitta, M.; Mitsumoto, Y.; Yamamoto, H. J. Chem. Soc., Perkin Trans. 1 2001, 1901.
Xiao, J.; Deng, L. Dalton Trans. 2013, 42, 5607. doi: 10.1039/c3dt50518a
Liu, J.; Hu, L.; Wang, L.; Chen, H.; Deng, L. J. Am. Chem. Soc. 2017, 139, 3876. doi: 10.1021/jacs.7b00484
Howell, J. A. S.; Palin, M. G.; McArdle, P.; Cunningham, D.; Goldschmidt, Z.; Gottlieb, H. E.; Hezroni-Langerman, D. Inorg. Chem. 1993, 32, 3493. doi: 10.1021/ic00068a019
宋礼成, 胡青眉, 周忠远, 胡国志, 向在筠, 无机化学学报, 1991, 7, 399. http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=19910406Song, L. C.; Hu, Q. M.; Zhou, Z. Y.; Hu, G. Z.; Xiang, Z. Y. Chin. J. Inorg. Chem. 1991, 7, 399 (in Chinese). http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=19910406
El-khateeb, M.; Al-Noaimi, M.; Al-Akhras, A.; Görls, H.; Weigan, W. J. Coord. Chem. 2012, 65, 2510. doi: 10.1080/00958972.2012.698406
Enemark, J. H.; Feltham, R. D.; Huie, B. T.; Johnson, P. L.; Swedo, K. B. J. Am. Chem. Soc. 1977, 99, 3285. doi: 10.1021/ja00452a015
Roesk, H. W.; Bertel, N.; Edelmann, F.; Noltemeyer, M.; Sheldrick G. M. Z. Naturforsch. 1988, 43b, 72.
Koketsu, J.; Ninomiya, Y.; Suzuki, Y.; Koga, N. Inorg. Chem. 1997, 36, 694. doi: 10.1021/ic951220u
Sudhakar, P. V.; Lammertsma, K. J. Am. Chem. Soc. 1991, 113, 1899. doi: 10.1021/ja00006a005
Sazama, G. T.; Betley, T. A. Organometallics 2011, 30, 4315. doi: 10.1021/om2003859
Hosokawa, S.; Ito, J.; Nishiyama, H. Organometallics 2012, 31, 8283. doi: 10.1021/om300901k
Peddarao, T.; Baishya, A.; Barman, M. K.; Kumara, A.; Nembenna, S. New J. Chem. 2016, 40, 7627. doi: 10.1039/C6NJ00907G
Chan, K. T. K.; Spencer, L. P.; Masuda, J. D.; McCahill, J. S. J.; Wei, P.; Stephan, D. W. Organometallics 2004, 23, 381. doi: 10.1021/om030539g
Betz, R.; Reichvilse, M. M.; ESchumi, E.; Miller, C.; Klüfers, P. Z. Anorg. Allg. Chem. 2009, 635, 1204. doi: 10.1002/zaac.v635:8
Olmstead, M. M.; Power, P. P.; Shoner, S. C. Inorg. Chem. 1991, 30, 2547. doi: 10.1021/ic00011a017
Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 4759. doi: 10.1002/anie.201001864
Evans, D. F. J. Chem. Soc. 1959, 2003. doi: 10.1039/jr9590002003
Sur, S. K. J. Magn. Reson. 1989, 82, 169.

图 1 磷亚胺和磷叶立德配位的亚铁配合物
Figure 1 Iminophosphorane-iron(Ⅱ) and phosphonium ylide- iron(Ⅱ) complexes

图式 1 高自旋砷亚胺配位的亚铁配合物4的合成路线
Scheme 1 Synthetic route to the high-spin iminoarsorane- iron(Ⅱ) complex 4

图 2 配合物3的分子结构(氢原子未显示)
Figure 2 Molecular structure of 3 with the omitting of hydrogen atom for clarity
Selected distances (nm) and angles (°): Fe(1)—N(1) 0.2038(7), Fe(1)—N(1A) 0.2038(7), Fe(1)—O(1) 0.2227(10), Fe(1)—O(2) 0.2168(10), Fe(1)—As(1) 0.2562(3), N(1)—Fe(1)—N(1A) 121.6(4), N(1)—Fe(1)—O(1) 117.9(2), N(1)—Fe(1)—O(2) 100.1(2), N(1)—Fe(1)—As(1) 81.3(2), As(1)—Fe(1)—O(1) 91.5(3), As(1)—Fe(1)—O(2) 177.0(3), O(1)—Fe(1)—O(2) 85.4(4)

图 3 配合物4分子结构(氢原子未显示)
Figure 3 Molecular structure of 4 with the omitting of hydrogen atom for clarity
Selected distances (nm) and angles (°): Fe(1)—N(1) 0.2008(3), Fe(1)—N(2) 0.2015(4), Fe(1)—N(3) 0.1996(3), As(1)—N(1) 0.1752(3), Fe(1)—O(1) 0.2112(4), N(1)—Fe(1)—N(2) 101.60(15), N(1)—Fe(1)—N(3) 99.58(14), N(2)—Fe(1)—N(3) 112.75(16), As(1)—N(1)—Fe(1) 98.45(16), N(1)—Fe(1)—O(1) 100.06(16)

图式 2 配合物4与2, 6-二甲基苯基异腈及三苯基膦的反应
Scheme 2 Reactions of 4 with ArNC (Ar=2, 6-dimethylphenyl) and PPh3

图 4 配合物5的分子结构(氢原子未显示)
Figure 4 Molecular structure of 5 with the omitting of hydrogen atom for clarity
Selected distances (nm) and angles (°): Fe(1)—N(1) 0.2052(3), Fe(1)—N(2) 0.2070(3), As(1)—Fe(1) 0.22927(6), Fe(1)—C(1) 0.1864(4), Fe(1)—C(2), 0.1811(4), Fe(1)—C(3) 0.1818(4), N(1)—Fe(1)—N(2) 91.5(1), N(1)—Fe(1)—C(1) 95.3(2), N(1)—Fe(1)—C(2) 90.7(1), N(1)—Fe(1)—C(3) 171.7(1), N(1)—Fe(1)—As(1) 83.02(9), N(2)—Fe(1)—C(1) 96.5(1), N(2)—Fe(1)—C(2) 174.5(1), N(2)—Fe(1)—C(3) 85.2(1), N(2)—Fe(1)—As(1) 79.19(9), As(1)—Fe(1)—C(1) 175.3(1), As(1)—Fe(1)—C(2) 96.1(1), As(1)—Fe(1)—C(3) 88.9(1)

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载: