引用本文:
姜海洋, 李强, 齐庆杰, 杨晨曦, 张丹. 金鸡纳碱衍生伯胺催化1-硝基-2-苯基乙烯与2-甲基丙醛不对称Michael加成反应机理的理论研究[J]. 有机化学,
2018, 38(4): 825-831.
doi:
10.6023/cjoc201703037 Citation:
Jiang Haiyang, Li Qiang, Qi Qingjie, Yang Chenxi, Zhang Dan. Theoretical Study on the Conjugate Addition of Asymmetric Michael Addition of trans-1-Nitro-2-phenylethylene to 2-Methylpropion-aldehyde Catalyzed by Cinchona Alkaloid Derived Primary Amine[J]. Chinese Journal of Organic Chemistry,
2018, 38(4): 825-831.
doi:
10.6023/cjoc201703037
Theoretical Study on the Conjugate Addition of Asymmetric Michael Addition of trans-1-Nitro-2-phenylethylene to 2-Methylpropion-aldehyde Catalyzed by Cinchona Alkaloid Derived Primary Amine
Received Date:
22 March 2017 Revised Date:
14 June 2017 Available Online:
25 April 2018
Fund Project:
Project supported by the China Postdoctoral Science Foundation Project (No. 2016M591451), the Natural Science Foundation of Liaoning Province (No. 2017054028), the Liaoning Education Department General Project (No. LJYL044), the Sixth Agricultural Technology Problems Foundation of Liaoning Technical University (No. 20160086T) and the Undergraduate Innovation and Entrepreneurship Training Program of Liaoning Province (No. 201610147000044)
Abstract:
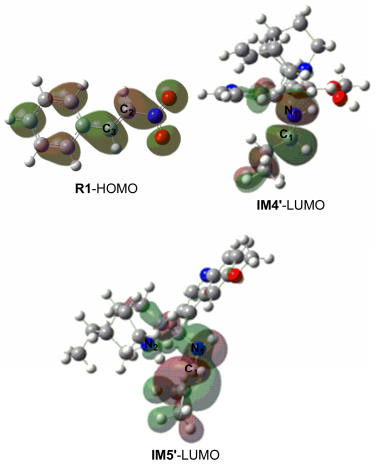
The theoretical study is presented for the Michael addition reaction between trans-1-nitro-2-phenylethylene and 2-methylpropionaldehyde catalyzed by (9S)-9-amino-6'-methoxy-10, 11-dihydrocinchonan (9-epi-DHQDA) and benzoic acid. All structures, including the reactants, intermediates, transition states and products were optimized. Transition states have been confirmed by the corresponding vibration analysis and intrinsic reaction coordinate (IRC). In addition, nature bond orbital (NBO) and atoms in molecules (AIM) theories have been used to analyze orbital interactions and bond natures. Calculations indicate that the benzoic acid might undergo a proton step to the 9-epi-DHQDA to produce the iminium intermediate. Then the iminium serves as a reactive acceptor to participate in the subsequent nucleophilic addition. Next, a proton transfer process from the tertiary amine to nitronate carbon is found to be rate-determining step, and the enantioselectivity of the catalyzed Michael reaction is also controlled by this step. Finally, one water molecule participates in hydrolysis and C=O bond formation, and results in the formation of product and recovery of catalyst.
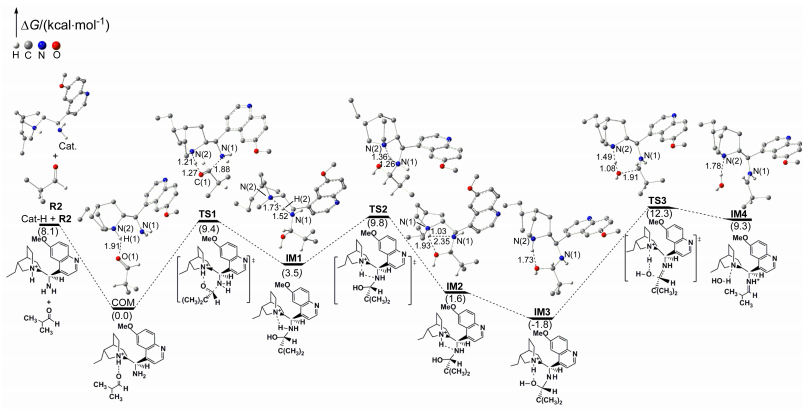
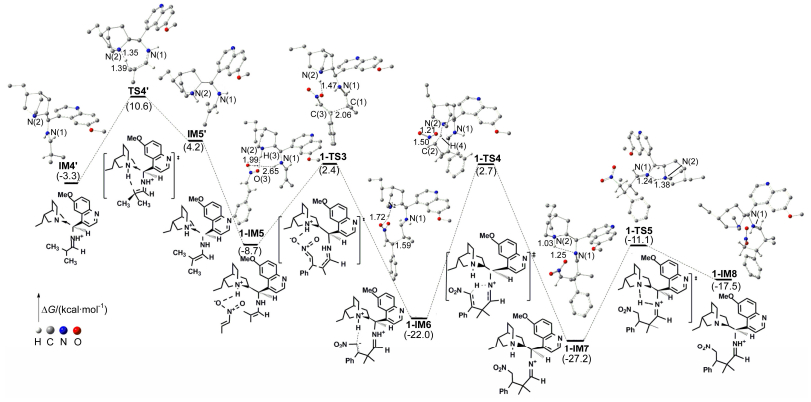
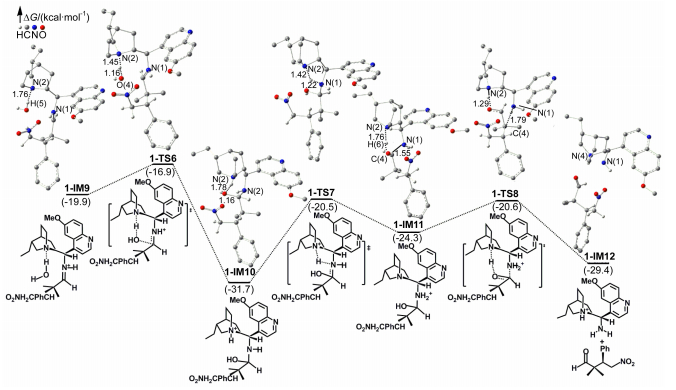
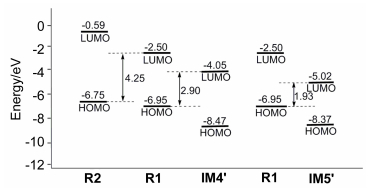
Figure 2.
Energy profile for the addition process along path 1 between iminium intermediate and trans-1-nitro-2-phenylethylene and the proton migration step along pathway 1
Some hydrogen atoms not involved in reaction sites are omitted. Distances are in Å. Values of ΔG are given in parentheses
McCooey, S. H.; Connon, S. J. Org. Lett.2007, 9, 599. doi: 10.1021/ol0628006
[36]
Gonzalez, C.; Schlegel, H. B. J. Chem. Phys.1989, 90, 2154. doi: 10.1063/1.456010
[37]
Gonzalez, C.; Schlegel, H. B. J. Phys. Chem. 1990, 94, 5523. doi: 10.1021/j100377a021
[38]
Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev.1988, 88, 899. doi: 10.1021/cr00088a005
[39]
Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys.1985, 83, 735. doi: 10.1063/1.449486
[40]
Frisch, M. J. ; Trucks, G. W. ; Schlegel, H. B. ; Scuseria, G. E. ; Robb, M. A. ; Cheeseman, J. R. ; Kudin, K. N. ; Burant, J. C. ; Millam, J. M. ; Iyengar, S. S. ; Tomasi, J. ; Barone, V. ; Mennucci, B. ; Cossi, M. ; Scalmani, G. ; Rega, N. ; Petersson, G. A. ; Nakatsuji, H. ; Hada, M. ; Ehara, M. ; Toyota, K. ; Fukuda, R. ; Hasegawa, J. ; Ishida, M. ; Nakajima, T. ; Honda, Y. ; Kitao, O. ; Nakai, H. ; Klene, M. ; Li, X. ; Knox, J. E. ; Hratchian, H. P. ; Cross, J. B. ; Adamo, C. ; Jaramillo, J. ; Gomperts, R. ; Stratmann, R. E. ; Yazyev, O. ; Austin, A. J. ; Cammi, R. ; Pomelli, C. ; Zakrzewski, V. G. ; Dapprich, S. ; Daniels, A. D. ; Strain, M. C. ; Farkas, O. ; Malick, D. K. ; Rabuck, A. D. ; Raghavachari, K. ; Foresman, J. B. ; Ortiz, J. V. ; Cui, Q. ; Baboul, A. G. ; Clifford, S. ; Piskorz, P. ; Komaromi, I. ; Martin, R. L. ; Fox, D. J. ; Keith, T. ; Challacombe, M. ; Johnson, B. ; Chen, W. ; Wong, M. W. ; Gonzalez. C. ; Pople, J. A. Gaussian 09, revision A. 1, Gaussian Inc., Wallingford CT, 2009.
[41]
Jones, G. O.; Li, X.; Hayden, A. E.; Houk, K. N.; Danishefsky, S. J. Org. Lett.2008, 10, 4093. doi: 10.1021/ol8016287
[42]
Peles, D. N.; Thoburn, J. D. J. Org. Chem.2008, 73, 3135. doi: 10.1021/jo702668u
[43]
Rehbein, J.; Hiersemann, M. J. Org. Chem.2009, 74, 4336. doi: 10.1021/jo900635k
Figure 2
Energy profile for the addition process along path 1 between iminium intermediate and trans-1-nitro-2-phenylethylene and the proton migration step along pathway 1
Some hydrogen atoms not involved in reaction sites are omitted. Distances are in Å. Values of ΔG are given in parentheses


 下载:
下载:

 下载:
下载:






