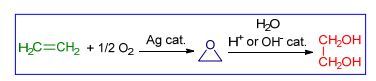
 图式1
环氧乙烷法合成乙二醇的反应路线
Scheme1.
Ethylene epoxide-mediated reaction route to ethylene glycol
图式1
环氧乙烷法合成乙二醇的反应路线
Scheme1.
Ethylene epoxide-mediated reaction route to ethylene glycol
引用本文:
张亦伟, 陈艺林, 方霄龙, 袁友珠, 朱红平. 草酸酯加氢制乙二醇钌金属均相催化体系的研究进展[J]. 有机化学,
2017, 37(9): 2275-2286.
doi:
10.6023/cjoc201703021

Citation: Zhang Yiwei, Chen Yilin, Fang Xiaolong, Yuan Youzhu, Zhu Hongping. Advances for the Ruthenium Complexes-Based Homogeneous Catalytic Hydrogenation of Oxalates to Ethylene Glycol[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2275-2286. doi: 10.6023/cjoc201703021
Citation: Zhang Yiwei, Chen Yilin, Fang Xiaolong, Yuan Youzhu, Zhu Hongping. Advances for the Ruthenium Complexes-Based Homogeneous Catalytic Hydrogenation of Oxalates to Ethylene Glycol[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2275-2286. doi: 10.6023/cjoc201703021
草酸酯加氢制乙二醇钌金属均相催化体系的研究进展
English
Advances for the Ruthenium Complexes-Based Homogeneous Catalytic Hydrogenation of Oxalates to Ethylene Glycol
Abstract:
Hydrogenation of oxalates is one of the important organic reactions, which has an ultimate use for the industrial production of ethylene glycol. The studies on the ruthenium complexes-based homogeneous catalytic reaction systems are herein summarized. With the focus on the catalytic reaction systems, the important factors with significant influences on the oxalate transformation efficiency as well as the product selectivity are discussed, including temperature, H2 pressure, catalyst concentration, reaction duration, additives, and so on. The catalytic reaction mechanisms are also discussed in detail, where the mechanism for the H2-heterolysis promoted under the metal-ligand cooperation for the oxalate hydrogenation to ethylene glycol is enhanced. This study would be useful for designing the new catalyst applicable in industry.
-
乙二醇(EG)是一种重要的基础有机化工原料, 用于防冻剂和溶剂.它也是精细化学品和药物合成用的原料以及聚酯和聚醚合成用的重要单体等[1].近年来95%的乙二醇用于生产聚对苯二甲酸乙二醇酯(PET). 2015年的统计数据表明全球乙二醇的产能为3100万吨, 产量达2700万吨, 而我国乙二醇的表观消费量接近1430万吨.至2030年, 全球对乙二醇产品的需求仍会保持一定的增长态势[2].近年来合成乙二醇的研究受到了很多科研工作者的关注, 聚焦点是工业化生产技术以及新型反应路线等.
乙二醇的合成路线主要包括以乙烯为原料的环氧乙烷法和以合成气为原料的草酸酯法.环氧乙烷法涉及乙烯在银催化剂的作用下与氧气发生环加成反应生成环氧乙烷, 以及环氧乙烷进一步在酸或碱催化剂的作用下水合转化成乙二醇(Scheme 1).目前该法的生产技术主要由英荷壳牌(Shell)公司、美国科学设计(SD)公司和陶氏化学(DOW)公司掌握[2].草酸酯法又称合成气氧化偶联法, 如Scheme 2所示, 第一步由甲醇、一氧化氮和氧气反应生成亚硝酸甲酯; 第二步亚硝酸甲酯与一氧化碳在钯催化剂作用下反应生成草酸二甲酯(DMO), 并释放出一氧化氮; 第三步草酸二甲酯在铜-铬或铜催化剂(注:工业用催化剂)作用下加氢转化成乙二醇, 并释放出甲醇.该路线由美国UCC公司于1966年提出, 1978年日本宇部兴产对该技术进行了改进, 近年来国内多家单位对该技术进行了革新, 并积极推向工业化生产[2].
图式1
环氧乙烷法合成乙二醇的反应路线
Scheme1.
Ethylene epoxide-mediated reaction route to ethylene glycol
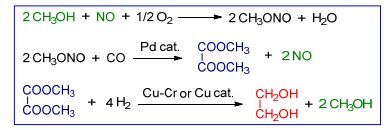 图式2
草酸酯法合成乙二醇的反应路线
Scheme2.
DMO-mediated reaction route to ethylene glycol
图式2
草酸酯法合成乙二醇的反应路线
Scheme2.
DMO-mediated reaction route to ethylene glycol
环氧乙烷法生产乙二醇是传统的石油化工路线, 其中原料乙烯依赖于石油的裂解.草酸酯法生产乙二醇则是现代煤化工路线.从Scheme 2的反应路线看, 一氧化氮和甲醇可以循环利用, 一氧化氮通过氨气和氧气反应制备, 氨气来源于合成气中的氢气与氮气经铁催化剂合成; 甲醇则可以经合成气工业生产.因此, 整个反应完全可以以合成气为原料.近年来, 合成气制烯烃产业得到很大发展, 其中产出的乙烯也可用于环氧乙烷法的原料, 也就是说环氧乙烷法也可以经煤化工路线实现.总的来说, 从化学原理角度看, 这两种路线都是热力学和动力学可行的反应, 并且原料来源丰富; 但从商业经济角度考虑, 这两种路线孰优孰劣则需要考察煤和石油资源的市场价格以及生产和技术成本.环氧乙烷法反应过程相对简单, 而草酸酯法则要复杂得多.后者的研究是近年来的热点.
在草酸酯法中, 一氧化碳与亚硝酸酯钯催化偶联是合成草酸酯的关键步骤, 但是草酸酯加氢是合成乙二醇的必要过程.草酸酯是一种特殊的双酯基碳碳键联的分子, 其加氢转化为醇是一种重要的酯类化合物的氢解反应[3-6].从分子反应历程看, 草酸酯需要与4个H2分子反应, 最后生成一个分子的乙二醇和两个分子的单醇, 这是一个非常复杂的反应过程.如Scheme 3所示, 草酸二甲酯中的一个酯基与H2在催化剂的作用下发生羰基键(C=O)加成生成半缩醛, 半缩醛不稳定重排脱去一个甲醇分子生成乙醛酸甲酯; 乙醛酸甲酯中的醛基进一步与H2在催化剂的作用下发生C=O键加成生成乙醇酸甲酯(MG).乙醇酸甲酯中的酯基经过类似的加氢过程生成乙二醇(EG).在整个反应过程中两种半缩醛分子以及乙醛酸甲酯和β-羟基乙醛均不稳定, 可看作是中间体; 但是乙醇酸甲酯是稳定的分子, 可以从反应中分离.因为草酸二甲酯和乙醇酸甲酯的加氢反应性有一定的差异性, 同时反应过程中常有副反应发生, 因此, 除了催化反应活性外, 反应选择性实际上也是评定催化反应体系好坏的一个重要因素, 这一点往往不被重视.
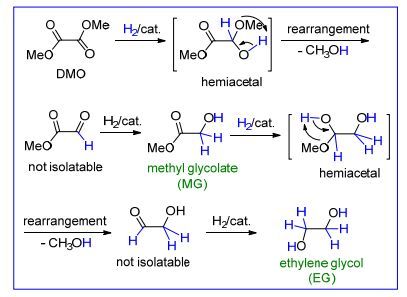 图式3
草酸二甲酯加氢生成乙二醇的基元反应过程
Scheme3.
Elemental reactions for hydrogenation of DMO to EG
图式3
草酸二甲酯加氢生成乙二醇的基元反应过程
Scheme3.
Elemental reactions for hydrogenation of DMO to EG
用于草酸酯加氢反应的催化剂主要包括无机物负载型金属催化剂和金属配合物催化剂两大类, 前者用于非均相反应, 后者则在均相反应体系中使用, 无论哪一种催化剂, 均需要经过如Scheme 3所示的反应历程.已经有一些文章对前者进行了综述讨论[7], 但是对后者的综述甚少.目前工业用催化剂集中于负载型, 但是由于负载型催化剂的复杂结构以及非均相反应特征, 研究提出的催化反应机理存在多种争论.相比较而言, 均相催化体系中的催化剂具有明确的结构组成, 而且整个体系反应条件温和, 易于实施, 可以通过多种测试手段对反应过程进行跟踪分析.这些使得相关反应机理的研究具有可行性和合理性.尽管如此, 针对Scheme 3所示的复杂转化, 化学家们至今对其中的反应机理尚无明确的定论.本文将从20世纪80年代草酸酯加氢钌金属均相催化反应体系的最初发现到目前的研究状况逐一论述, 重点介绍典型的催化剂体系, 并探讨反应条件对产物的影响以及催化反应机理.
1 草酸酯加氢制备乙二醇的钌金属均相催化体系
早期的研究显示酯类分子加氢生成醇主要使用氢化试剂, 如氢化锂铝、硼氢化钠等, 反应后需要进一步水解形成产物, 但是该过程要消耗计量的氢化试剂, 同时产生金属氢氧化物或氧化物或盐等废弃物[3~6, 8~9].因此这种方法在初期探索方面有一定的积极意义, 但不具有应用价值.负载型的金属催化剂, 如二氧化硅负载的铜-铬、铜等相继被发现, 但是加氢反应条件苛刻, 如早期报道的反应温度达到200~250 ℃, 压力达到13.0~21.3 MPa[10, 11].目前的研究表明通过添加助剂、或调变金属组分的比例、以及优化催化剂的微环境结构等, 可使反应条件温和很多.
1980年, 美国联合化学公司的Grey和Pez研究小组[12a, 12b]合成了膦配体稳定的钌氢离子型化合物1和2(表 1).他们在探索醛、酮、碳酸酯以及腈类分子催化加氢反应后, 首次拓展至草酸二甲酯底物, 但是反应活性并不理想.以四氢呋喃(THF)为溶剂, 在90 ℃和620 kPa氢压下, 催化剂1在20 h内催化生成10%的乙醇酸甲酯(Entry 1), 2可以催化得到70%的产率(Entry 2).当反应体系中加入18-冠醚-6试图络合稳定钾离子, 提高钌中心对底物分子的转换频率, 但是结果并不好, 产率反而降为53% (Entry 3).作者在文中只是提及草酸二甲酯是一种活性酯, 活性程度可类比于三氟甲基乙酸酯, 但没有讨论反应机理.不过他们在引言中述及, 对于如1和2型的催化剂, 反应过程可能经历阴离子部分M—H键对C=O键的加成, 再进一步与H2作用并解离转换形成产物, 活性组分还原.其中MH2的生成是H2基团交换的结果还是钌中心氧化加成的结果未有提及, 这一点实际上对揭示催化剂作用的本质非常重要. A正离子对C=O氢化反应后成盐作用显而易见, 这也可以解释为什么加入18-冠醚-6反而会引起转化率的下降, 因为18-冠醚-6络合A离子降低了成盐作用(Scheme 4).尽管如此, 这种推测的作用方式与上述氢化试剂的Meerwein-Ponndorf-Verley反应过程不同[12c, 12d].
 表 1
钌氢化物阴离子催化DMO加氢转化为MGa
Table 1.
Transformation of DMO to MG catalyzed by the ruthenium hydride anion
表 1
钌氢化物阴离子催化DMO加氢转化为MGa
Table 1.
Transformation of DMO to MG catalyzed by the ruthenium hydride anion
Entry Cat. Yield/% MG EG 1 1 10 0 2 2 70 0 3 2/18-crown-6 53 0 aSolvent: THF, p(H2): 620 kPa, temperature: 90 ℃, time: 20 h. 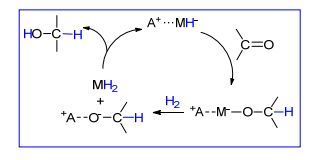 图式4
Grey和Pez建议的金属氢化物离子催化丙酮加氢反应的可能机理
Scheme4.
Grey and Pez proposed mechanism for hydrogenation of acetone catalyzed by the ruthenium hydride anion
图式4
Grey和Pez建议的金属氢化物离子催化丙酮加氢反应的可能机理
Scheme4.
Grey and Pez proposed mechanism for hydrogenation of acetone catalyzed by the ruthenium hydride anion
1984年, Matteoli课题组[13]也对草酸酯加氢催化进行了研究.他们合成了中性钌羰基氢化物3和4(表 2), 随后考察了催化性能.在180 ℃、13.2 MPa H2压、苯溶剂中, 化合物3在144 h内催化51.1%草酸二甲酯生成乙醇酸甲酯(Entry 1);当催化剂量增加1倍时, 反应在39 h内完成, 转化率达100% (Entry 2), 但是4没有活性. 3和4的差别仅是所用膦配体不同, 但两者有着如此大的差异, 作者没有深究, 也没有探讨相关反应机理.值得注意的是, 该反应条件与负载型催化剂的接近.于是他们进一步以碳负载的5为催化剂, 在相同条件下考察表明, 反应活性非常低, 转化率仅为3.0% (Entry 4).
表 2
钌氢化物催化DMO加氢转化为MGa
Table 2.
Ruthenium hydride-catalyzed transformation of DMO to MG
Entry Cat. t/h Yield/% MG EG 1 3b 144 51.1 0 2 3c 39 100 0 3 4 144 0.0 0 4 5d 144 3.0 0 aSolvent: benzene, p(H2): 13.2 MPa, temperature: 180 ℃; bamount of 0.05 g; camount of 0.10 g; d amount of 0.05 g/1 g (C). 在3的催化反应中, H4Ru4(CO)9(PBu3)3和H4Ru4-(CO)10(PBu3)2物种被检测到, 表明在反应过程中发生了膦配体的解离, 因而促进了低配位活性钌中心的形成.碳负载金属钌催化剂通过H4Ru4(CO)12在碳上热分解制备, 该催化剂显示极低活性, 显然膦配体较碳载体具更好的配位稳定作用, 有利于活性中心的形成.催化剂3对草酸二乙酯也显示反应活性, 如在Entry 4的条件下, 草酸二乙酯的转化率为37.7%, 但是产物很复杂.
1985年, 该研究小组[15]报道了系列含膦配体的羰基钌乙酸化合物以及催化性能研究(表 3).仍在13.2 MPa氢压、180 ℃、苯溶剂中, 三苯基膦配位的钌化合物11的反应活性与上述结果类似, 仍然极低(144 h后生成6.0%乙醇酸甲酯, Entry 20), 相比较而言, 其它膦配体羰基钌化合物均显示较好活性.在该条件下, 多核化合物6可以转化51.5%的草酸二甲酯为乙醇酸甲酯(Entry 1), 而双核化合物10的转化效率为41.3% (Entry 19).化合物7~9的结果随反应条件的变化而变化.
表 3
钌羰基化合物催化DMO加氢转化为MG和EGa
Table 3.
Ruthenium carbonyl-catalyzed transformation of DMO to MG and EG
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% MG EG 1b 6 13.2 180 144 51.5 0 2b 7 13.2 180 144 79.2 0 3 7 13.2 180 2 97.9 2.1 4 7 13.2 180 22 13.4 0 5 7 13.2 180 72 61.6 38.4 6 7 13.2 120 72 96.1 3.9 7 8 13.2 180 2 70.1 4.3 8 8 13.2 180 22 71.1 0 9 8 13.2 180 72 1.9 32.6 10 8 13.2 150 72 15.3 21.0 11 8 13.2 135 72 54.1 10.2 12 8 13.2 120 72 72.3 27.7 13 8 20.3 120 72 40.5 59.5 14 8 1.0 120 72 17.2 6.1 15 9 13.2 180 72 7.7 11.7 16 9 13.2 180 2 66.3 0.8 17 9 13.2 180 22 29.2 0 18 9 13.2 120 72 81.9 18.1 19b 10 13.2 180 144 41.3 0 20b 11 13.2 180 144 6.0 0 21 12 13.2 180 2.50 100 0 22 13 13.2 180 72 28.7 66.5 aSolvent: MeOH; b benzene. 针对催化剂7, 在保持13.2 MPa氢压和180 ℃下苯(144 h后乙醇酸甲酯的得率为79.2%, Entry 2) 和甲醇溶剂(22 h后的得率为13.4%, Entry 4) 对反应有很大影响; 进一步以甲醇为溶剂, 温度和反应时间对转化率也显示影响(120 ℃、72 h后生成96.1%的乙醇酸甲酯和3.9%的乙二醇, Entry 6; 180 ℃、2 h时生成97.9%的乙醇酸甲酯和2.1%的乙二醇, Entry 3, 72 h后生成61.8%的乙醇酸甲酯和31.4%的乙二醇, Entry 5).这些数据表明乙醇酸甲酯主要是低温下的反应产物, 升高温度则促进乙二醇的生成, 但同时导致产物分解, 如生成甲酸甲酯、二氧化碳和甲烷等.因此, 高温长时间内测得的数据实际上是产物分解后的数据.催化剂8和9的结果与7相比具体数值有一定差异, 但受温度和时间的影响类似, 其中8的结果也同时受到H2压的影响.
单核化合物12和13也具有催化活性(表 3).以12为催化剂, 控制13.2 MPa氢气压和180 ℃反应温度, 在甲醇中草酸二甲酯氢化时0~2.5 h内几乎只得到乙醇酸甲酯和甲醇, 反应呈0.5级(在苯溶剂中呈1级反应特征); 延长反应时间, 另一种产物乙二醇逐步生成, 但是不具有任何动力学转化的特征.一方面催化剂的活性逐渐降低, 反应转化率也随之降低; 另一方面出现其它副产物如二氧化碳、乙酸甲酯、二(2-羟乙基)醚和乙醇酸-2-羟基乙酯等.
1991年, 他们[15]报道13可将草酸二甲酯选择性地还原为乙二醇.为避免底物的分解, 氢化反应需要在120 ℃下进行, 促进草酸二酯完全转化, 之后再在180 ℃下将中间体乙醇酸甲酯氢化.
总的来说, 催化剂6~13对产物的选择性受温度控制, 但是过高温度引起产物的分解; 同时可以看出不同膦配体的催化剂结果有差异.具体的催化反应机理未有讨论.
1997年, Elsevier课题组[16]选择乙酰丙酮钌(Ru(acac)3)作为金属前驱体, 与系列配体组成催化剂体系考察了草酸二甲酯的加氢反应. Hara和Wada课题组[17]已经研究了乙酰丙酮钌/配体催化酸酐氢化为內酯的反应, 反应在甲醇中进行, 同时需要金属锌的参与.他们认为锌促进乙酰丙酮钌的快速还原, 原位产生催化活性中心(表 4).在100 ℃、7.1 MPa氢压、甲醇溶剂中, 单独使用乙酰丙酮钌/锌基本上没有活性(41 h, 2% MG, Entry 1);加入配体AsPh3、PCy3、邻菲罗啉双氮L21、三氮L22以及吡唑硼氢钾盐L23也均无活性.相比较而言, 其它单齿、双齿和多齿配体的加入能够催化反应的进行.以16 h为计量时间, 加入PPh3、L17、L18、L19分别生成36%、11%、67%、85%的乙醇酸甲酯(Entries 2和5~7), 而加入三脚架型膦配体L20会同时生成乙醇酸甲酯和乙二醇, 两者的量随锌使用量变化, 在该反应条件下最好的结果是锌的加入量为20%时, 草酸二甲酯的转化率达100%, 乙二醇的生成量为95% (Entry 8).
表 4
不同配体-钌化合物催化DMO加氢转化为MG和EG的反应结果a
Table 4.
Results for hydrogenation of DMO to MG and EG catalyzed by Ru complexes incorporating varied ligands
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% MG EG 1 Ru(acac)3/Zn 7.0 100 41 2 0 2 Ru(acac)3/L14/Zn 7.0 100 16 36 0 3 Ru(acac)3/L15/Zn 7.0 100 16 0 0 4 Ru(acac)3/L16/Zn 7.0 100 16 1 0 5 Ru(acac)3/L17/Zn 7.0 100 16 11 0 6 Ru(acac)3/L18/Zn 7.0 100 16 67 0 7 Ru(acac)3/L19/Zn 7.0 100 16 85 0 8 Ru(acac)3/L20/Znb 7.0 100 16 1 95 9 Ru(acac)3/L20/Znc 7.0 100 16 15 52 10 Ru(acac)3/L20/Zn 3.8 100 16 24 10 11 Ru(acac)3/L20/Zn 2.0 100 16 19 2 12 Ru(acac)3/L20 7.0 100 16 24 22 13 Ru(acac)3/L20/Znd 2.0 100 16 0 84 14 Ru(acac)3/L20c 2.0 100 16 10 63 15 Ru(acac)3/L21/Zn 7.0 100 16 0 0 16 Ru(acac)3/L22/Zn 7.0 100 16 0 0 17 Ru(acac)3/L23/Zn 7.0 100 16 1 0 aSolvent: MeOH; b22% Zn; c24% Zn; din dry MeOH. 明显地可以看出, 配体对金属钌的络合稳定对催化活性起着非常重要的作用, 膦配体的稳定作用要远好于氮配体; 在膦配体中稳定作用顺序大致为: PCy3<Ph2PC2H4PPh2<PPh3<PhP(C2H4PPh2)2≈[CH2P(Ph)-C2H4PPh2]2≪MeC(CH2PPh2).
相比于Matteoli课题组的工作, Elsevier课题组的催化剂体系活性高, 且反应条件温和, 其中关键是多齿膦配体的使用. MeC(CH2PPh2)3配体能够与钌中心形成“口袋式”配位, 给钌中心与底物分子间的作用提供了所要求的电子和立体空间, 这一点非常必要.基于温和的反应条件, 副反应以及产物分解反应也得到了一定程度的遏制.
对Ru(acac)3/L20/Zn体系, 不论提升压力还是增加催化剂浓度均能提高草酸二甲酯的转化率(单位时间的转换频率也增加), 但是Zn的加入并不是决定因素(无Zn参与仍显示催化活性, 见Entries 12和14), 在相同条件下加入Zn总体上会提高反应活性.
2003年, 他们[18]将工作延伸至其它金属前驱体, 发现使用RuCl2(PPh3)4, 在120 ℃、8.1 MPa氢压下甲醇溶剂中17 h内, 草酸二甲酯的转化率为51%, 乙醇酸甲酯的收率仅为20%, 没有乙二醇生成; 而在THF溶剂中草酸二甲酯的转化率为37%和乙醇酸甲酯的收率为12%.原位混合的RuCl3/PPh3给出44%的转化率和15%的收率, 同样没有乙二醇产生.以RuCl2(PPh3)4为前驱体的体系如何转化生成活性氢化物物种至今尚不明确.
2006年, Hanton研究小组[19]报道了一种三脚架型含硫配体MeC(CH2SBu)3 (L27), 并研究了与Ru(acac)3组成的催化剂体系用于草酸二甲酯的选择性氢化.含硫配体已经被报道与Rh、Pd、Ir和Pt形成络合物, 用于烯烃和酮类分子的加氢催化, 但是从未用于酯类分子[20].在100 ℃、8.0 MPa氢压、甲醇溶剂中, Hanton研究小组考察证实没有膦配体参与的Ru(acac)3无活性, 该结果印证了Elsevier组的考察结果(表 5).因而加入POct3反应具有活性, 但需要304 h完成草酸二甲酯完全转化为乙醇酸甲酯(Entry 2), 加入PPh3在5.7 h后全部转化为乙二醇(Entry 1).尽管反应条件中H2压微有差异, 但是后者的结果与Elsevier组的相差很大(表 5 Entry 1 vs表 4 Entry 2).当使用硫环配体时, L25和L26没有促进作用, 而三脚架型L31显示很好的作用, 随着反应时间从23 h增加到136 h, 乙醇酸甲酯的收率从36.5%增加到100% (表 5 Entries 5~7), 但没有乙二醇生成.在前两个反应中, 可以观察到“钌黑”的生成, 表明L25和L26并不能有效地配位稳定钌中心, 导致钌被还原为金属态.在使用L27的所有反应中, 打开高压釜仍然观察到黄色透明溶液, 没有任何沉淀.这说明三脚架型硫配体在这些条件下具有超强的络合稳定Ru(Ⅱ)的能力.
表 5
不同配体-钌化合物催化DMO加氢转化为MG和EG的反应结果
Table 5.
Results for hydrogenation of DMO to MG and EG catalyzed by Ru complexes incorporating varied ligands
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% MG EG 1 Ru(acac)3/L18/Zn 8.0 100 5.7 0 100 2 Ru(acac)3/L24/Zn 8.0 100 304 100 0 3 Ru(acac)3/L25/Zn 8.0 100 20 0 0 4 Ru(acac)3/L26/Zn 8.0 100 20 0 0 5 Ru(acac)3/L27/Zn 8.0 100 23 36.9 0 6 Ru(acac)3/L27/Zn 8.0 100 69 87.2 0 7 Ru(acac)3/L27/Zn 8.0 100 136 100 0 8 Ru(acac)3/L28/Zn 8.0 100 20.9 96.6 0 9 Ru(acac)3/L28/Zn 6.0 100 66 35.9 64.1 10 Ru(acac)3/L28/Zn 7.0 100 84 58.2 41.8 11 Ru(acac)3/L28/Zn 8.0 100 66 46.5 53.5 12 Ru(acac)3/L28/Zn 11.0 100 34 97.3 2.7 13 Ru(acac)3/L28/Zn 14.0 100 86 80.2 19.8 14 Ru(acac)3/L28/Zn 8.0 80 82 80.9 18.1 15 Ru(acac)3/L28/Zn 8.0 90 61 46.9 53.1 16 Ru(acac)3/L28/Zn 8.0 110 64 60.0 40.0 17 Ru(acac)3/L28/Zn 8.0 120 41 58.2 41.8 18 Ru(acac)3/L29/Zn 8.0 100 20.6 93.0 5.6 aSolvent: MeOH. 添加和不添加Zn的催化体系均需要一段时间才能观察到反应的发生, 这预示着Ru(Ⅲ)前驱体在反应中转化为Ru(Ⅱ)活性物种需要时间, 即存在一个诱导期, 锌的添加的确有助于缩短诱导期, 因而反应速率得到提高.相比于三脚架型膦配体, 三脚架型硫配体只能将草酸二甲酯还原为乙醇酸甲酯, 且与反应时间无关.在新鲜配制的催化剂溶液中直接加入乙醇酸甲酯底物, 也没有任何转化.由此可见不同配体影响着钌的催化能力.进一步研究发现Ru(acac)3/L27/Zn和Ru(acac)3/L18/Zn体系体系对底物分子都呈零级反应, 说明草酸二甲酯的浓度对反应无影响.
2011年, Hanton研究小组[21]考察了Ru(acac)3/L18体系添加Zn以及其它有机化合物的影响(图 1).他们首先证实该反应体系存在诱导期, 即将催化组分和底物混合后需要一段时间才观察到反应的进行. Zn的加入可以将反应诱导期从160 min缩短到30 min, 同时反应时间也会从10.1 h减少到5.7 h.但是加入如图 1所示的有机分子添加剂(30, 1, 5, 7-三氮杂二环[4.4.0]-5-癸烯; DAE, 二异丙基氨基乙醇; DMAP, 4-二甲氨基吡啶; 4NP, 4-硝基苯酚), 诱导期时间会缩短, 不过反应时间均会拉长, 同时产物在数和量上也会变化.对吡啶、L30、DMAP添加剂, 仅生成产物乙醇酸甲酯; 对苯酚, 得到乙二醇; 而其它添加剂则会导致生成乙醇酸甲酯和乙二醇的混合物, 且百分占比不同.显然这些有机物添加剂对草酸二甲酯氢化速率并没有起到促进作用.推测这些添加剂有可能以两种不同的方式作用, 一种是通过电子给体的作用协助催化活性物种的生成; 另一种则是与底物作用.推测当后者起主导作用时, 大大影响了催化反应速率.
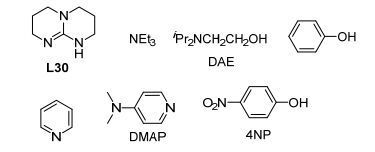 图1
Hanton组用于Ru(acac)3/L18体系的有机添加剂
Figure1.
Additives added in Ru(acac)3/L18 system reported by Hanton group
图1
Hanton组用于Ru(acac)3/L18体系的有机添加剂
Figure1.
Additives added in Ru(acac)3/L18 system reported by Hanton group
在酯的氢化反应中, 工业界特别关注的是催化体系对有机羧酸杂质的耐受度, 许多多相催化技术往往受制于这些杂质, 因为它们会引起催化剂的流失.为此, Hanton研究组研究了Ru(acac)3/L18/Zn体系受羧酸的影响.他们在加入草酸二甲酯的同时也加入10 mol%的乙醇酸, 在100 ℃、8.0 MPa氢压、甲醇溶剂中, 经过60 min的诱导期和12 h的反应时间, 100%的反应物转化为乙二醇. GC-MS没有检测到乙醇酸.其它实验也表明乙醇酸在甲醇中60 ℃下完全转化为乙醇酸甲酯, 因此加入的乙醇酸应该先酯化然后被氢化, 再转化为乙二醇.
强σ-电子配位的膦配体是很适合的稳定钌金属的配体, 呈多齿配位特性的配体则更佳.配体L27和L28有类似于L20的三脚架型的配位特征, 同时N原子也会参与金属的配位, 因此这两个配体配位的钌化合物是很好的酯加氢催化剂. Hanton组证实在100 ℃、8.0 MPa氢压、甲醇溶剂中, 在20 h内Ru(acac)3/L27/Zn催化草酸二甲酯生成96.6%的乙醇酸甲酯(表 5 Entry 8), 而Ru(acac)3/L28/Zn催化生成93.0%的乙醇酸甲酯和5.6%的乙二醇(表 5 Entry 18).动力学考察表明这两个反应都对底物倾向于一级, 即反应速率受草酸二甲酯的浓度影响.推测在反应过程中草酸二甲酯与钌中心发生强的络合, 该络合步骤是反应的决速步.尽管氢压的变化对产物乙醇酸甲酯和乙二醇的生成比例有影响(较低氢压对生成乙二醇有利而高压对乙醇酸甲酯有利(表 5 Entries 9~13), 但是对反应速率影响甚微, 显然H2分子在钌中心作用形成金属氢活性物种并不包含在决速步内.该推测与动力学观察到的对草酸二甲酯为一级反应的结果相符.温度变化对反应影响似乎不大, 在80 ℃下反应十分缓慢, 以10 ℃为步长从90 ℃升温至120 ℃, 反应速率没有明显变化.普遍认为催化剂会随着温度升高而加剧H2的氧化加成形成活性物种, 同时也促进对酯羰基的加成, 当然也会容易引起催化剂的分解.很可能是升高温度带来的速率提升与催化剂分解导致催化剂浓度降低而引起的速率降低相抵消, 使得总反应表观速率变化不大.
依据上述系列结果, 他们提出一种可能的反应机理.如Scheme 5所示, Ru(acac)3/L27/Zn经过诱导期反应, 脱除戊二醇生成三脚架型膦配体配位的RuⅡH2物种. GC谱分析到痕量的2, 4-戊二醇予以证实.这是一个钌中心16电子结构的化合物, 易于与底物草酸二甲酯络合配位. PPh3 (L18)配体体系观察到对底物呈零级, 表明钌与草酸二甲酯结合与解离都很容易.然而L27配体体系对底物呈一级, 显然由L27配位的钌络合草酸二甲酯容易而解离难, 该络合反应是决速步.钌氢插入羰基后容易形成RuCO2四元环结构, 再经还原消除生成半缩醛分子和[L27]Ru(0) 物种, [L27]Ru(0) 与H2氧化加成重新还原为[L27]RuⅡH2用于下一个催化反应, 半缩醛分子则自发重排为乙醛酸甲酯, 在类似的反应过程中被氢化为乙醇酸甲酯.乙二醇分子则是由乙醇酸甲酯经过相应的催化循环反应生成.
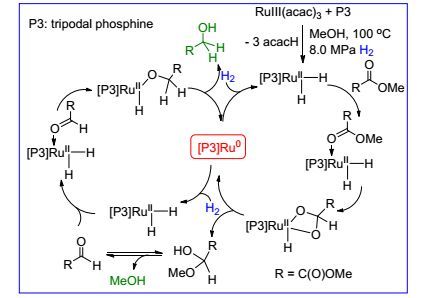 图式5
Hanton推测的反应机理
Scheme5.
Mechanism suggested by Hanton group
图式5
Hanton推测的反应机理
Scheme5.
Mechanism suggested by Hanton group
2013年, Beller课题组[22]考察了如表 6所示的系列催化剂对草酸酯加氢的催化性能.他们首先使用Takasago公司首创的钳型Macho钌催化剂31[23].在100 ℃、6.0 MPa H2、四氢呋喃溶剂和NaOEt存在下, 该催化剂在20 h内催化草酸二乙酯直接转化为乙二醇, 收率为92% (Entry 1).在相同条件下使用氯离子被氢负离子BH3加合物替代的催化剂32, 给出略高的转化率(96%, Entry 2);控制时间1 h, 反应生成21%的乙醇酸乙酯和72%的乙二醇(Entry 3).该结果说明反应经过乙醇酸乙酯中间体, 在较长的反应时间内其完全转化为乙二醇.
表 6
不同催化体系催化DMO加氢转化为乙醇酸乙酯和EG的反应结果
Table 6.
Results for hydrogenation of DMO to ethyl glycolate and EG with different catalyst systems
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% Ethyl glycolate EG 1 31/NaOEt 6.0 100 20 0 92 2 32/NaOEt 6.0 100 20 0 96 3 32/NaOEt 6.0 100 1 21 72 4 32 6.0 100 1 15 85 5 32 6.0 100 4 0 >99 6 32 6.0 60 1 99 0 7 32 6.0 r.t. 120 7 86 8 32 3.0 60 1 46 0 9b 32 6.0 60 0.5 98 0 10c 32 9.0 60 1 0 0 11d 32 9.0 60 1 0 0 12e 32 7.0 120 140 81 0 13 33/NaOEt 6.0 100 20 89 0 14 34/NaOEt 6.0 100 20 96 0 15 L35/Ru(acac)3/NaOEt 6.0 100 20 0 0 16 L36/Fe(BF4)2/NaOEt 6.0 100 20 0 0 17 L36/Co(BF4)2/NaOEt 6.0 100 20 0 0 18 L37/Ru(acac)3/NaOEt 6.0 100 20 0 0 19 {[L37]FeF}BF4/NaOEt 6.0 100 20 0 0 20 L38/[Ru(acac)3]/NaOEt 6.0 100 20 0 0 21f L20/Ru(acac)3/NaOEt 6.0 100 20 0 96 aSolvent: THF; bEtOH as solvent; ctoluene as solvent; dpropylene carbonate as solvent; esolvent free; fMeOH as solvent. 在不使用NaOEt的情况下, 32也具有很好的催化活性, 在1 h内转化草酸二乙酯生成15%的乙醇酸乙酯和85%的乙二醇(Entry 4), 4 h后得到大于99%的乙二醇(Entry 5).当降至60 ℃时, 1 h内生成99%的乙醇酸乙酯(Entry 6);而室温下120 h内给出7%的乙醇酸乙酯和86%的乙二醇(Entry 7).同样氢压和溶剂变换也对催化反应有影响, 在60 ℃、3.0 MPa H2、四氢呋喃溶剂中1 h下仅生成46%的乙醇酸乙酯(Entry 8);而在乙醇溶剂中、60 ℃、6.0 MPa H2、0.5 h内生成98%的乙醇酸乙酯(Entry 9);但是使用甲苯或碳酸丙烯酯溶剂, 没有活性(Entries 10和11).无溶剂参与时, 在120 ℃、7.0 MPa H2、140 h内只能产生81%的乙醇酸乙酯(Entry 12).
使用NaOEt可以促进钌中心的卤素基团转化为钌烷氧基, 进而转化为氢负离子活性基团, 这一点已为很多课题组的研究证实.因此, 上述结果表明32可以看成是真正的催化活性物种.
变换31和32中的PPh2为PiPr2(催化剂33和34), 相应的反应速率变化不大, 但是只生成乙醇酸乙酯(Entries 13和14).可以注意到催化剂31~34中的配体含有NH配位基, 因此Beller课题组进一步研究发现不包含NH基的多齿膦配体(L35~L37)、膦氮配体(L38)以及氮配体(39)与金属组成的化合物在与上述几乎相同的反应条件下(100 ℃、6.0 MPa H2、四氢呋喃溶剂和NaOEt)没有任何反应活性(Entries 15~20).然而L20/ Ru(acac)3/NaOEt体系在MeOH溶剂中显示活性, 催化草酸二甲酯生成96%的乙二醇.这些结果表明质子性溶剂对催化反应的引发相当重要, 可能是促进活性物种的形成, 但是文中没有深入的讨论.催化剂32对草酸二甲酯和二丁酯也都有很好的反应活性, 前者底物在1 h、100 ℃下被转化得到84%的乙二醇, 而后者底物在3 h、60 ℃下被转化为95%的乙醇酸丁酯.
2013年, Elsiever课题组[24]合成了离子型化合物40, 并考察了催化性能.在80 ℃、8.0 MPa H2、四氢呋喃溶剂、KOiBu和24 h下催化草酸二甲酯生成29%的乙醇酸甲酯, 催化活性不佳.
从上述的综述结果看出, 草酸酯加氢存在两种主要的催化反应机理. Grey和Hanton课题组都推测氢气在钌中心作用下生成RuH2, 然后RuH2中的一个氢负离子与底物分子发生加成, 由此产生的基团与另外一个氢负离子再还原消除.在这个过程中钌中心氧化态经历0→+Ⅱ的循环变化过程.该过程被认为是金属中心主导的“内核作用过程”(inner sphere interaction process)[25]. Beller课题组的研究结果则确证配体NH以及质子性溶剂的重要作用. Noyori[26]、Morris[27]、Bergens[28]、Saudan[29]、丁奎岭[30]等课题组在其它羰基分子加氢反应的研究中提出金属-NH配体的协同作用原理, 称为“外部作用过程”(outer sphere proton-hydride transfer process, 经过“六元环”的过渡态).与前者相比, 该过程不涉及钌中心氧化态的变化.同时可以看出, 在这个过程中H2发生了异裂反应.但是迄今为止尚没有人给出确凿的实验证据证明该过程.
2014年周其林课题组[31]合成了一种空气稳定的双吡啶四齿配体新型钌化合物41, 该化合物在温和条件下高效催化一系列脂肪酯和芳香酯的加氢还原, 催化剂使用量最低仅有0.001 mol%.在5.1 MPa氢压、25 ℃、异丙醇溶剂中, 反应16 h后草酸二甲酯转化生成乙醇酸异丙醇酯.反应需要加入NaOMe.进一步升高反应温度至100 ℃, 反应完全转化为乙二醇.他们推测在NaOMe和H2气氛中, 41转化生成钌氢活性物种, 与底物分子中的C=O键加成, 再通过“膦臂”CH2基团质子氢转移促进单醇分子的消除; 随后醛基经过类似的氢化过程转化为醇基.该反应机理与Milstein课题组[32]和Song课题组[33]报道的结果类似, 相应的41被认为是一种“金属-配体”双功能化作用的催化剂.
近年来我们在非均相催化草酸二甲酯为乙醇酸甲酯和乙二醇方面开展了多方面的研究工作[34]. 2016年, 我们将研究从非均相拓展到均相催化反应体系, 合成了膦氮配体钌化合物催化剂, 用于草酸二甲酯的加氢反应.在该研究中, 我们选择含NH基的PN螯合型刚性配体与钌组成催化体系, 详细探讨了催化剂的合成和性能(表 7)[35].
表 7
PN配体钌催化体系催化DMO加氢转化为MG的反应结果a
Table 7.
Results for hydrogenation of DMO to MG catalyzed by PN-ligand coordinated ruthenium catalysts
Entry Cat. T/℃ t/h Yield/% MG EG 1 RuCl2(PPh3)3/NaOMe 100 1 0 0 2 RuCl2(PPh3)3/o-PPh2-C6H4NH2/NaOMe 100 48 97 0 3 RuCl2(PPh3)3/2o-PPh2-C6H4NH2/NaOMe 100 96 97 0 4 RuCl2(PPh3)3/3o-PPh2-C6H4NH2/NaOMe 100 120 73 0 5 42/NaOMe 100 1 97 0 6 43/NaOMe 100 1 97 0 7 43/NaOMec 25 16 98 0 8 44/NaOMe 100 1 0 0 9 45 100 20 92 0 10 45/NaOMe 100 20 96 0 11 47 100 3 86 0 12 47/NaOMe 100 3 99 0 aSolvent: THF, NaOMe: 10 equiv, DMO: 200 equiv, p(H2): 5.1 MPa. c 2000 equiv. DMO. RuCl2(PPh3)3/o-PPh2C6H4NH2/NaOMe体系在100℃、5.1 MPa H2、四氢呋喃溶剂中催化草酸二甲酯转化为乙醇酸甲酯, 但是结果受金属与配体比例的影响.当为1:1时, 在48 h内产率为97%; 1:2时96 h内得到相同结果; 而1:3时即使反应120 h也只得到73%的产率.这些结果暗示不同比例的反应体系可能生成不同结构的催化活性物种.
为验证这种推测, 在催化反应温度下我们以甲苯为溶剂分别进行了RuCl2(PPh3)3与o-PPh2C6H4NH2按上述计量比的反应, 在12 h后得到单配体配位的化合物42, 而48 h后得到双配体配位的化合物43.当使用RuHCl(PPh3)3为前驱体时, 在24 h后得到三配体配位的离子型化合物44.相应的性能考察表明, 42/NaOMe和43/NaOMe在上述条件下1 h内转化生成97%的乙醇酸甲酯(43/NaOMe也能够催化2000 equiv.的底物在16 h后得到98%的乙醇酸甲酯, Entry 7), 但是44/NaOMe没有任何活性.这些结果显示原位组成的体系需要一定的时间形成PN配体配位的钌化合物, 进而生成活性物种.上述RuCl2(PPh3)3/3o-PPh2C6H4NH2/NaOMe体系的催化结果表明反应过程应该经历42和43的形成, 并逐步转化为44, 最后致使反应停止, 因而底物分子不能完全转化.
我们选择42与K[HBsBu3]反应得到了钌氢化合物45(图 2).同时合成了另一种双配体配位的钌化合物46, 其与K[HBsBu3]反应得到另一种钌氢化合物48.该化合物加热至70 ℃时脱去一分子氢气转化成47.这样在无NaOMe的参与下45和47均能直接催化草酸二甲酯转化为乙醇酸甲酯[产率分别是92% (20 h内)和86% (3 h内)]; 添加NaOMe则提高产率至96%和99%.
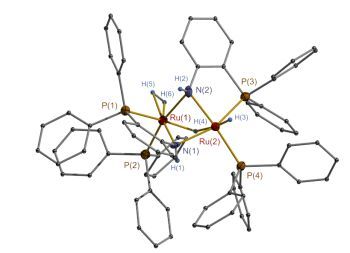 图2
化合物45的单晶结构
Figure2.
X ray crystal structure of catalyst 45
图2
化合物45的单晶结构
Figure2.
X ray crystal structure of catalyst 45
化合物47和48可以相互转化(Eq. 1, 图 3), 不难发现H2分子的脱除源自RuH和配体中的NH2基团的作用, 即两基团从键接状态HRu←NH2变为RuNH(后者易于二聚), 而RuNH与H2反应又转回HRu←NH2键接状态(注: Noyori等课题组将这种转换定义为Ru—N单键和双键之间的变化.我们采用单键和配位键的变化意在表明Ru中心的氧化态保持不变, 与“内核作用原理”不同). D2实验显示这种转化即使在10 ℃时仍然进行.
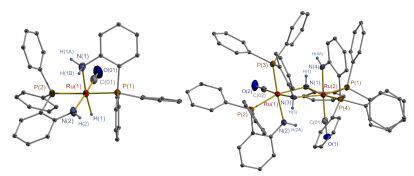 图3
化合物48(左)和47(右)的单晶结构
Figure3.
X-ray crystal structures of complexes 48 (left) and 47 (right)
图3
化合物48(左)和47(右)的单晶结构
Figure3.
X-ray crystal structures of complexes 48 (left) and 47 (right)
这样, 我们对上述催化反应机理进行了推测.如Scheme 6所示, 处于HRu←NH2键接状态的催化活性物种48与底物分子中的C=O通过六元环过渡态的电性作用引发氢负离子-质子转移, 产生半缩醛分子, 其重排脱去一分子的甲醇生成乙醛酸甲酯; 该分子中醛基C=O再与48经上述作用生成乙醇酸甲酯产物.氢负离子-质子迁移后的钌物种易于二聚形成47, 47在反应过程中可看成是一种“稳态”, 其进一步与H2反应重新形成活性态48.
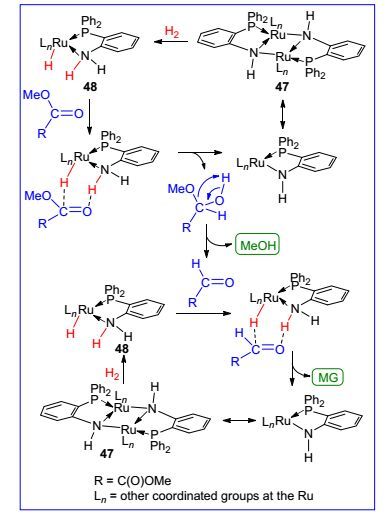 图式6
推测的反应机理
Scheme6.
Suggested mechanism
图式6
推测的反应机理
Scheme6.
Suggested mechanism
进一步研究发现调变反应条件时, 42/NaOMe和43/NaOMe都能催化草酸二甲酯到乙二醇.以上述反应条件为基础, 提高一倍NaOMe量、升高温度至120 ℃、DMO量减半时, 42在36 h后催化生成31%的乙醇酸甲酯和61%的乙二醇; 在72 h后生成25%的乙醇酸甲酯和68%的乙二醇; 进一步升高温度至140 ℃在36 h后使乙醇酸甲酯完全转化得到94%的乙二醇.使用催化剂43, 在120 ℃、36 h后乙醇酸甲酯转化完全得到97%的乙二醇.
表 8
PN配体钌催化体系催化DMO加氢转化为MG和EG的反应结果a
Table 8.
Results for hydrogenation of DMO to MG and EG catalyzed by PN-ligand coordinated ruthenium catalysts
Entry Cat. T/℃ t/h Yield/% MG EG 1 42/20 NaOMe 120 36 31 61 2 42/20 NaOMe 120 72 25 68 3 42/20 NaOMe 140 36 0 94 4 43/20 NaOMe 120 16 51 47 5 43/20 NaOMe 120 24 20 75 6 43/20 NaOMe 100 36 0 97 aSolvent: THF, DMO: 100 equiv., p(H2): 5.1 MPa. 显而易见, 使用同一种钌化合物42或43作催化剂在催化得到乙二醇时需要更为苛刻的条件.这也印证了草酸二甲酯的反应活性要强于乙醇酸甲酯.升高反应温度是促进直接转化成乙二醇的一种有效途径, 但是使用更多量的NaOMe亦可显著提高乙二醇的生成.值得注意的是, 该反应是在封闭体系中进行, 甲醇是体系中的副产物.从动力学角度考虑, 生成乙二醇会产生两个分子的甲醇.这样生成乙二醇的反应要比生成乙醇酸甲酯的难很多.对比也会发现, 使用同一种催化剂, 转化为乙二醇产物的时间要比转化为乙醇酸甲酯的时间长得多.乙二醇的生成机理类似于Scheme 6, 只是将其中草酸二甲酯底物换为乙醇酸甲酯.在含NH基团的PN配体钌化合物催化剂体系中, 我们确定了重要化合物48和47的结构, 揭示了这两个化合物之间内含的H2分子转换, 证实了它们的催化性能, 这些为“金属-NH配体”协同的“外部催化作用机理”提供了可靠的实验证据.
3 总结与展望
草酸酯的钌金属均相催化加氢反应是一个明确的溶液均相态的分子反应过程, 其中关键的步骤涉及到H2分子的活化进而加成到底物分子中的C=O上.使用配体钌化合物为催化剂, 配体必需具有强的配位能力来稳定钌中心发生官能化作用.对于“外部氢负离子-氢质子转移作用过程”, 配体包含协同金属中心异裂H2的基团为佳, 否则要使用质子性的溶剂来协同金属中心作用.反应条件对反应的进行和转化相当重要.温度决定分子碰撞程度, 升高温度有利于反应速率的提高, 但是过高的温度也会引起产物的分解以及其它副反应.氢压决定H2分子在溶液中的浓度, 高H2压有利于反应向产物方向的动力学转化.碱性添加剂如醇钠会促进钌中心特别是含氯基团生成活性氢负离子基, 同时它也会对配体中含N基团的亲核性有重要影响[28].使用不同酯底物时, 醇钠有选择性, 如对于草酸二甲酯, 通常使用甲醇钠, 而对于草酸二乙酯则使用乙醇钠等.金属锌添加剂在针对乙酰丙酮钌前驱体时, 具有很好地促进三价钌还原的作用.
在这个反应体系中, 乙醇酸甲酯和乙二醇都是重要的目标产物.对决大多数反应来说, 同一种催化剂都具有转化生成这两种产物的催化能力, 但是由于草酸酯和乙醇酸甲酯的反应性差异, 乙醇酸甲酯生成比较容易, 进一步转化成乙二醇需要调变反应条件, 这个调变需要在保证产物不分解的前提下才有意义.从反应工艺上讲, 目前的催化反应多在反应釜密闭体系中进行, 体系中会产生单醇副产物, 从反应体系中移出产物和副产物将会大大提高反应效率.如能在反应工艺中实现这个操作, 均相催化反应体系将不失为一个极具潜力的应用体系, 因为该体系反应条件要较非均相体系温和很多, 而且反应效率高, 并能有效地控制乙醇酸甲酯和乙二醇的高选择性产出.
-
-
[1]
(a) Jiang, Z. Synthetic Technology and Application 2010, 25(4), 27(in Chinese).
(江镇海, 合成技术及应用, 2010, 25(4), 27. )
(b) Huang, G. ; Li, X. ; Yang, Y. ; Qu, J. Petrochem. Technol. Appl. 2015, 33, 75(in Chinese).
(黄格省, 李雪静, 杨延翔, 曲静波, 石化技术与应用, 2015, 33, 75. ) -
[2]
(a) Li, D. ; Wang, H. Modern Chem. Ind. 2017, 37(1), 5(in Chinese).
(李代红, 王洪波, 现代化工, 2017, 37(1), 5. )
(b) Liu, Z. Chem. Ind. Eng. Proc. 2013, 32, 1214(in Chinese).
(刘宗语, 化工进展, 2013, 32, 1214. )
(c) Chen, W. ; Sun, J. ; Zhang, J. ; Zhang, S. ; Hua, W. Chem. Ind. Eng. Proc. 2014, 33, 1740(in Chinese).
(成卫国, 孙剑, 张军平, 张锁江, 华炜, 化工进展, 2014, 33, 1740. ) -
[3]
(a) Gaylord, N. G. J. Chem. Educ. 1957, 34, 367.
(b) Hudlicky, M. Reductions in Organic Chemistry, ACS, Washington, DC, 1996. -
[4]
(a) Modler, R. F.; Gubler, R.; Inoguchi, Y. Detergent Alchols, CEH Marketing Research Report, 2004.
(b) Hong, W. S. Natural Detergent Alcohols by a Vapour Phase Ester Hydrogenation Process, PPP Review 93-2-1, 2004.
(c) Cant, N. W.; Trimm, D. L.; Turek, T. Catal. Rev. Sci. Eng. 1994, 36, 645. -
[5]
March, J. Advanced Organic Chemistry:Reactions Mechanisms and Structure, 4th ed., Wiley-Interscience, New York, 1992, p. 1213.
-
[6]
(a) Rylander, P. N. Catalytic Hydrogenation in Organic Syntheses, Academic Press, New York, 1979.
(b) de Vries, J. G.; Elsevier, C. J. Handbook of Heterogeneous Hydrogenation for Organic Synthesis, Weily-VCH, Weinheim, 2007. -
[7]
(a) Yue, H. -R. ; Zhao, Y. -J. ; Ma, X. -B. ; Gong, J. -L. Chem. Soc. Rev. 2012, 41, 4218.
(b) Zhao, Y. ; Zhao, S. ; Wang, B. ; Lv, J. ; Ma, X. Chem. Indu. Eng. Prog. 2013, 32, 721(in Chinese).
(赵玉军, 赵硕, 王博, 吕静, 马新宾, 化工进展, 2013, 32, 721. )
(c) Jin, E. -L. ; Zhang, Y. -L. ; He, L. -L. ; Harris, H. G. ; Teng, B. -T. ; Fan, M. -H. Appl. Catal. A: Gen. 2014, 476, 158.
(d) Higman, C. ; Tam, S. Chem. Rev. 2014, 114, 1673.
(e) Li, J. ; Duan, X. ; Lin, H. ; Ye, L. ; Yuan, Y. Petrochem. Technol. 2014, 43, 985(in Chinese).
(李建辉, 段新平, 林海强, 叶林敏, 袁友珠, 石油化工, 2014, 43, 985. )
(f) Li, S. -M. ; Wang, Y. ; Zhang, J. ; Wang, S. -P. ; Xu, Y. ; Zhao, Y. -J. ; Ma, X. -B. Ind. Eng. Chem. Res. 2015, 54, 1243. -
[8]
(a) Bayón, J. C.; Claver, C.; Masdeu-Bultó, A. M. WO 2003093208, 2003[Chem. Abstr. 2003, 139, 366612].
(b) Pope, S. J. A.; Champness, N. R.; Reid, G. J. Chem. Soc., Dalton Trans. 1997, 1639. -
[9]
Seyden-Penne, J. Reductions by the Allumino-and Borohydride in Organic Synthesis, 2nd ed.; Wiley-VCH, New York, 1997.
-
[10]
(a) Turek, T.; Trimm, D.; Cant, N. Catal. Rev. -Sci. Eng. 1994, 36, 645.
(b) Pouilloux, Y.; Autin, F.; Barrault, J. Catal. Today 2000, 63, 87. -
[11]
Pritchard, J.; Filonenko, G. A.; van Putten, R.; Hensen, E. J. M.; Pidko, E. A. Chem. Soc. Rev. 2015, 44, 3808. doi: 10.1039/C5CS00038F
-
[12]
(a) Grey, R. A.; Pez, G. P.; Wallo, A.; Corsi, J. J. Chem. Soc., Chem. Commun. 1980, 783.
(b) Grey, R. A.; Pez, G. P.; Wallo, A. J. Am. Chem. Soc. 1981, 103, 7536.
(c) De Graauw, C. F.; Peters, J. A.; van Bekkum, H.; Huskens, J. Synthesis 1994, 1007.
(d) Ashby, E. C. Acc. Chem. Res. 1988, 21, 414. -
[13]
Matteoli, U.; Bianchi, M.; Menchi, G.; Frediani, P.; Piacenti, F. J. Mol. Catal. 1984, 22, 353. doi: 10.1016/0304-5102(84)80075-9
-
[14]
(a) Matteoli, U.; Bianchi, M.; Menchi, G.; Frediani, P.; Piacenti, F. J. Mol. Catal. 1985, 29, 269.
(b) Matteoli, U.; Menchi, G.; Bianchi, M.; Piacenti, F. J. Organomet. Chem. 1986, 299, 233.
(c) Matteoli, U.; Bianchi, M.; Menchi, G.; Piacenti, F. J. Mol. Catal. 1988, 44, 347. -
[15]
(a) Matteoli, U.; Bianchi, M.; Menchi, G.; Piacenti, F. J. Mol. Catal. 1991, 64, 257.
(b) Matteoli, U.; Menchi, G.; Bianchi, M.; Piacenti, F.; Ianelli, S.; Nardelli, M. J. Organomet. Chem. 1995, 498, 177. -
[16]
Teunissen, H. T.; Elsevier, C. J. Chem. Commun. 1997, 667.
-
[17]
Kara, Y.; Wada, K. Chem. Lett. 1991, 20, 553. doi: 10.1246/cl.1991.553
-
[18]
van Engelen, M. C.; Teunissen, H. T.; de Vries, J. G.; Elsevier, C. J. J. Mol. Catal. A:Chem. 2003, 206, 185. doi: 10.1016/S1381-1169(03)00427-8
-
[19]
Boardman, B.; Hanton, M. J.; van Rensburg, H.; Tooze, R. P. Chem. Commun. 2006, 2289.
-
[20]
Bayón, J. C.; Claver, C.; Masdeu-Bultó, A. M. Coord. Chem. Rev. 1999, 193~195, 73.
-
[21]
Hanton, M. J.; Tin, S.; Boardman, B. J.; Miller, P. J. Mol. Catal. A:Chem. 2011, 346, 70. doi: 10.1016/j.molcata.2011.06.010
-
[22]
Ziebart, C.; Jackstell, R.; Beller, M. ChemCatChem 2013, 5, 3228. doi: 10.1002/cctc.201300209
-
[23]
Kuriyama, W.; Matsumoto, T.; Ogata, O.; Ino, Y.; Aoki, K.; Tanaka, S.; Ishida, K.; Kobayashi, T.; Sayo, N.; Saito, T. Org. Process Res. Dev. 2012, 16, 166. doi: 10.1021/op200234j
-
[24]
Eveline, J.; Jongbloed, L. S.; Tromp, D. S.; Martin, L.; Bas, D. B.; Elsevier, C. J. ChemSusChem 2013, 6, 1737. doi: 10.1002/cssc.201300363
-
[25]
(a) Bryndza, H. E.; Tam, W. Chem. Rev. 1988, 88, 1163.
(b) Pàmies, O.; Bäckvall, J. E. Chem. Eur. J. 2001, 7, 5052.
(c) Fulton, J. R.; Holland, A. W.; Fox, D. J.; Vergman, R. G. Acc. Chem. Res. 2002, 35, 44.
(d) Clapham, S. E.; Hadzovic, A.; Morris, R. H. Coord. Chem. Rev. 2004, 248, 2201. -
[26]
(a) Yamakawa, M.; Ito, H.; Noyori, R. J. Am. Chem. Soc. 2000, 122, 1466.
(b) Noyori, R.; Ohkuma, T. Angew. Chem. Int. Ed. 2001, 40, 40.
(c) Ohkuma, T.; Koizumi, M.; Muñiz, K.; Hilt, G.; Kabuto, C.; Noyori, R. J. Am. Chem. Soc. 2002, 124, 6508.
(d) Sandoval, C. A.; Ohkuma, T.; Muñiz, K.; Noyori, R. J. Am. Chem. Soc. 2003, 125, 13490. -
[27]
(a) Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A.; Harvey, J. N.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2002, 124, 15104.
(b) Abdur-Rashid, K.; Faatz, M.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2001, 123, 7473.
(c) Abbel, R.; Abdur-Rashid, K.; Faatz, M.; Hadzovic, A.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2005, 127, 1870.
(d) Abdur-Rashid, K.; Guo, R.; Lough, A. J.; Morris, R. H.; Song, D. Adv. Synth. Catal. 2005, 347, 571. -
[28]
(a) Hamilton, R. J.; Bergens, S. H. J. Am. Chem. Soc. 2006, 128, 13700.
(b) Hamilton, R. J.; Bergens, S. H. J. Am. Chem. Soc. 2008, 130, 11979.
(c) Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H. J. Am. Chem. Soc. 2011, 133, 9666.
(d) John, J. M.; Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H. J. Am. Chem. Soc. 2013, 135, 8578. -
[29]
Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P. Angew. Chem., Int. Ed. 2007, 46, 7473. doi: 10.1002/(ISSN)1521-3773
-
[30]
(a) Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Angew. Chem., Int. Ed. 2012, 51, 13041.
(b) Zhao, B.; Han, Z.; Ding, K. Angew. Chem., Int. Ed. 2013, 52, 4744. -
[31]
Li, W.; Xie, J.; Yuan, M.; Zhou, Q. Green Chem. 2014, 16, 4081. doi: 10.1039/C4GC00835A
-
[32]
Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. Angew. Chem., Int. Ed. 2006, 45, 1113. doi: 10.1002/(ISSN)1521-3773
-
[33]
(a) Sun, Y.; Koehler, C.; Tan, R.; Annibale, V. T.; Song, D. Chem. Commun. 2011, 47, 8349.
(b) Yang, X. ACS Catal. 2012, 2, 964. -
[34]
(a) He, Z.; Lin, H.; He, P.; Yuan, Y. J. Catal. 2011, 277, 54.
(b) Wang, Y.; Duan, X.; Zheng, J.; Lin, H.; Yuan, Y.; Ariga, H.; Takakusagi, S.; Asakura, K. Catal. Sci. Technol. 2012, 2, 1637.
(c) Huang, Y.; Ariga, H.; Zheng, X.; Duan, X.; Takakusagi, S.; Asakura, K.; Yuan, Y. J. Catal. 2013, 307, 74.
(d) Zheng, J.; Lin, H.; Wang, Y.; Zheng, X.; Duan, X.; Yuan, Y. J. Catal. 2013, 297, 110.
(e) Zheng, X.; Lin, H.; Zheng, J.; Duan, X.; Yuan, Y. ACS Catal. 2013, 3, 2738.
(f) Li, M.; Ye, L.; Zheng, J.; Fang, H.; Kroner, A.; Yuan, Y.; Tsang, S. C. E. Chem. Commun. 2016, 52, 2569.
(g) Li, M.; Zheng, J.; Qu, J.; Liao, F.; Raine, E.; Kuo, W. C. H.; Su, S. S.; Po, P.; Yuan, Y.; Tsang, S. C. E. Sci. Rep. 2016, 6, 20527.
(h) Zheng, J.; Duan, X.; Lin, H.; Gu, Z.; Fang, H.; Li, J.; Yuan, Y. Nanoscale 2016, 8, 5959. -
[35]
Fang, X.; Zhang, C.; Chen, J.; Zhu, H.; Yuan, Y. RSC Adv. 2016, 6, 45512. doi: 10.1039/C6RA00320F
-
[1]
-

图式1 环氧乙烷法合成乙二醇的反应路线
Scheme 1 Ethylene epoxide-mediated reaction route to ethylene glycol

图式4 Grey和Pez建议的金属氢化物离子催化丙酮加氢反应的可能机理
Scheme 4 Grey and Pez proposed mechanism for hydrogenation of acetone catalyzed by the ruthenium hydride anion

图 1 Hanton组用于Ru(acac)3/L18体系的有机添加剂
Figure 1 Additives added in Ru(acac)3/L18 system reported by Hanton group

图 3 化合物48(左)和47(右)的单晶结构
Figure 3 X-ray crystal structures of complexes 48 (left) and 47 (right)
表 1 钌氢化物阴离子催化DMO加氢转化为MGa
Table 1. Transformation of DMO to MG catalyzed by the ruthenium hydride anion
Entry Cat. Yield/% MG EG 1 1 10 0 2 2 70 0 3 2/18-crown-6 53 0 aSolvent: THF, p(H2): 620 kPa, temperature: 90 ℃, time: 20 h.  下载: 导出CSV
下载: 导出CSV
表 2 钌氢化物催化DMO加氢转化为MGa
Table 2. Ruthenium hydride-catalyzed transformation of DMO to MG
Entry Cat. t/h Yield/% MG EG 1 3b 144 51.1 0 2 3c 39 100 0 3 4 144 0.0 0 4 5d 144 3.0 0 aSolvent: benzene, p(H2): 13.2 MPa, temperature: 180 ℃; bamount of 0.05 g; camount of 0.10 g; d amount of 0.05 g/1 g (C).
下载: 导出CSV
表 3 钌羰基化合物催化DMO加氢转化为MG和EGa
Table 3. Ruthenium carbonyl-catalyzed transformation of DMO to MG and EG
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% MG EG 1b 6 13.2 180 144 51.5 0 2b 7 13.2 180 144 79.2 0 3 7 13.2 180 2 97.9 2.1 4 7 13.2 180 22 13.4 0 5 7 13.2 180 72 61.6 38.4 6 7 13.2 120 72 96.1 3.9 7 8 13.2 180 2 70.1 4.3 8 8 13.2 180 22 71.1 0 9 8 13.2 180 72 1.9 32.6 10 8 13.2 150 72 15.3 21.0 11 8 13.2 135 72 54.1 10.2 12 8 13.2 120 72 72.3 27.7 13 8 20.3 120 72 40.5 59.5 14 8 1.0 120 72 17.2 6.1 15 9 13.2 180 72 7.7 11.7 16 9 13.2 180 2 66.3 0.8 17 9 13.2 180 22 29.2 0 18 9 13.2 120 72 81.9 18.1 19b 10 13.2 180 144 41.3 0 20b 11 13.2 180 144 6.0 0 21 12 13.2 180 2.50 100 0 22 13 13.2 180 72 28.7 66.5 aSolvent: MeOH; b benzene.
下载: 导出CSV
表 4 不同配体-钌化合物催化DMO加氢转化为MG和EG的反应结果a
Table 4. Results for hydrogenation of DMO to MG and EG catalyzed by Ru complexes incorporating varied ligands
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% MG EG 1 Ru(acac)3/Zn 7.0 100 41 2 0 2 Ru(acac)3/L14/Zn 7.0 100 16 36 0 3 Ru(acac)3/L15/Zn 7.0 100 16 0 0 4 Ru(acac)3/L16/Zn 7.0 100 16 1 0 5 Ru(acac)3/L17/Zn 7.0 100 16 11 0 6 Ru(acac)3/L18/Zn 7.0 100 16 67 0 7 Ru(acac)3/L19/Zn 7.0 100 16 85 0 8 Ru(acac)3/L20/Znb 7.0 100 16 1 95 9 Ru(acac)3/L20/Znc 7.0 100 16 15 52 10 Ru(acac)3/L20/Zn 3.8 100 16 24 10 11 Ru(acac)3/L20/Zn 2.0 100 16 19 2 12 Ru(acac)3/L20 7.0 100 16 24 22 13 Ru(acac)3/L20/Znd 2.0 100 16 0 84 14 Ru(acac)3/L20c 2.0 100 16 10 63 15 Ru(acac)3/L21/Zn 7.0 100 16 0 0 16 Ru(acac)3/L22/Zn 7.0 100 16 0 0 17 Ru(acac)3/L23/Zn 7.0 100 16 1 0 aSolvent: MeOH; b22% Zn; c24% Zn; din dry MeOH.
下载: 导出CSV
表 5 不同配体-钌化合物催化DMO加氢转化为MG和EG的反应结果
Table 5. Results for hydrogenation of DMO to MG and EG catalyzed by Ru complexes incorporating varied ligands
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% MG EG 1 Ru(acac)3/L18/Zn 8.0 100 5.7 0 100 2 Ru(acac)3/L24/Zn 8.0 100 304 100 0 3 Ru(acac)3/L25/Zn 8.0 100 20 0 0 4 Ru(acac)3/L26/Zn 8.0 100 20 0 0 5 Ru(acac)3/L27/Zn 8.0 100 23 36.9 0 6 Ru(acac)3/L27/Zn 8.0 100 69 87.2 0 7 Ru(acac)3/L27/Zn 8.0 100 136 100 0 8 Ru(acac)3/L28/Zn 8.0 100 20.9 96.6 0 9 Ru(acac)3/L28/Zn 6.0 100 66 35.9 64.1 10 Ru(acac)3/L28/Zn 7.0 100 84 58.2 41.8 11 Ru(acac)3/L28/Zn 8.0 100 66 46.5 53.5 12 Ru(acac)3/L28/Zn 11.0 100 34 97.3 2.7 13 Ru(acac)3/L28/Zn 14.0 100 86 80.2 19.8 14 Ru(acac)3/L28/Zn 8.0 80 82 80.9 18.1 15 Ru(acac)3/L28/Zn 8.0 90 61 46.9 53.1 16 Ru(acac)3/L28/Zn 8.0 110 64 60.0 40.0 17 Ru(acac)3/L28/Zn 8.0 120 41 58.2 41.8 18 Ru(acac)3/L29/Zn 8.0 100 20.6 93.0 5.6 aSolvent: MeOH.
下载: 导出CSV
表 6 不同催化体系催化DMO加氢转化为乙醇酸乙酯和EG的反应结果
Table 6. Results for hydrogenation of DMO to ethyl glycolate and EG with different catalyst systems
Entry Cat. p(H2)/MPa T/℃ t/h Yield/% Ethyl glycolate EG 1 31/NaOEt 6.0 100 20 0 92 2 32/NaOEt 6.0 100 20 0 96 3 32/NaOEt 6.0 100 1 21 72 4 32 6.0 100 1 15 85 5 32 6.0 100 4 0 >99 6 32 6.0 60 1 99 0 7 32 6.0 r.t. 120 7 86 8 32 3.0 60 1 46 0 9b 32 6.0 60 0.5 98 0 10c 32 9.0 60 1 0 0 11d 32 9.0 60 1 0 0 12e 32 7.0 120 140 81 0 13 33/NaOEt 6.0 100 20 89 0 14 34/NaOEt 6.0 100 20 96 0 15 L35/Ru(acac)3/NaOEt 6.0 100 20 0 0 16 L36/Fe(BF4)2/NaOEt 6.0 100 20 0 0 17 L36/Co(BF4)2/NaOEt 6.0 100 20 0 0 18 L37/Ru(acac)3/NaOEt 6.0 100 20 0 0 19 {[L37]FeF}BF4/NaOEt 6.0 100 20 0 0 20 L38/[Ru(acac)3]/NaOEt 6.0 100 20 0 0 21f L20/Ru(acac)3/NaOEt 6.0 100 20 0 96 aSolvent: THF; bEtOH as solvent; ctoluene as solvent; dpropylene carbonate as solvent; esolvent free; fMeOH as solvent.
下载: 导出CSV
表 7 PN配体钌催化体系催化DMO加氢转化为MG的反应结果a
Table 7. Results for hydrogenation of DMO to MG catalyzed by PN-ligand coordinated ruthenium catalysts
Entry Cat. T/℃ t/h Yield/% MG EG 1 RuCl2(PPh3)3/NaOMe 100 1 0 0 2 RuCl2(PPh3)3/o-PPh2-C6H4NH2/NaOMe 100 48 97 0 3 RuCl2(PPh3)3/2o-PPh2-C6H4NH2/NaOMe 100 96 97 0 4 RuCl2(PPh3)3/3o-PPh2-C6H4NH2/NaOMe 100 120 73 0 5 42/NaOMe 100 1 97 0 6 43/NaOMe 100 1 97 0 7 43/NaOMec 25 16 98 0 8 44/NaOMe 100 1 0 0 9 45 100 20 92 0 10 45/NaOMe 100 20 96 0 11 47 100 3 86 0 12 47/NaOMe 100 3 99 0 aSolvent: THF, NaOMe: 10 equiv, DMO: 200 equiv, p(H2): 5.1 MPa. c 2000 equiv. DMO.
下载: 导出CSV
表 8 PN配体钌催化体系催化DMO加氢转化为MG和EG的反应结果a
Table 8. Results for hydrogenation of DMO to MG and EG catalyzed by PN-ligand coordinated ruthenium catalysts
Entry Cat. T/℃ t/h Yield/% MG EG 1 42/20 NaOMe 120 36 31 61 2 42/20 NaOMe 120 72 25 68 3 42/20 NaOMe 140 36 0 94 4 43/20 NaOMe 120 16 51 47 5 43/20 NaOMe 120 24 20 75 6 43/20 NaOMe 100 36 0 97 aSolvent: THF, DMO: 100 equiv., p(H2): 5.1 MPa.
下载: 导出CSV
-










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 2560
- HTML全文浏览量: 368
 下载:
下载:
