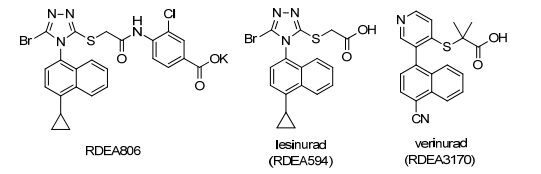
 图 1
抗病毒药物RDEA806及URAT1抑制剂lesinurad和verinurad的结构
Figure 1.
Structures of antiviral agent RDEA806 and URAT1 inhibitors lesinurad and verinurad
图 1
抗病毒药物RDEA806及URAT1抑制剂lesinurad和verinurad的结构
Figure 1.
Structures of antiviral agent RDEA806 and URAT1 inhibitors lesinurad and verinurad
引用本文:
田禾, 吴景卫, 刘钰强, 谢亚非, 王建武, 赵桂龙. 含烷氧基取代的三唑类结构的尿酸转运体1抑制剂的高效合成方法[J]. 有机化学,
2017, 37(7): 1748-1756.
doi:
10.6023/cjoc201701038

Citation: Tian He, Wu Jingwei, Liu Yuqiang, Xie Yafei, Wang Jianwu, Zhao Guilong. Efficient Synthetic Approaches to Uric Acid Transporter 1 Inhibitors Bearing Alkoxyl Group-Substituted Triazoles[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1748-1756. doi: 10.6023/cjoc201701038
Citation: Tian He, Wu Jingwei, Liu Yuqiang, Xie Yafei, Wang Jianwu, Zhao Guilong. Efficient Synthetic Approaches to Uric Acid Transporter 1 Inhibitors Bearing Alkoxyl Group-Substituted Triazoles[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1748-1756. doi: 10.6023/cjoc201701038
含烷氧基取代的三唑类结构的尿酸转运体1抑制剂的高效合成方法
摘要:
含烷氧基取代的三唑类结构的尿酸转运体1(URAT1)抑制剂3-(4-(4-环丙基萘-1-基)-5-甲氧基-4H-1,2,4-三唑-3-基)丙酸(1a)和3-[4-(4-环丙基萘-1-基)-5-乙氧基-4H-1,2,4-三唑-3-基]丙酸(1b)是一类重要的药物先导化合物,但是其现有合成路线收率非常低(1a和1b的总收率分别为3.3%和3.0%),为了对其进行进一步的构效关系研究,需要收率高的路线.经过详细研究发现了两条高效的合成路线(A和B),分别以CuCl催化的醇钠对溴代三唑进行芳香族亲核取代反应和醇钠直接对甲磺酰基取代的三唑进行芳香族亲核取代反应作为关键反应,并对重要步骤的反应条件进行深入优化.这两条路线具有收率高的优点,除了可以作为先导化合物1a和1b继续进行构效关系研究的合成路线外,还可以为含烷氧基取代的其他杂环化合物的合成提供有价值的借鉴.
English
Efficient Synthetic Approaches to Uric Acid Transporter 1 Inhibitors Bearing Alkoxyl Group-Substituted Triazoles
Abstract:
Uric acid transporter 1 (URAT1) inhibitors bearing alkoxy group-substituted triazoles 3-(4-(4-cyclopropylnaphthalen-1-yl)-5-methoxy-4H-1, 2, 4-triazol-3-yl)propanoic acid (1a) and 3-(4-(4-cyclopropylnaphthalen-1-yl)-5-ethoxy-4H-1, 2, 4-triazol-3-yl)propanoic acid (1b) are structurally interesting lead compounds in drug design. The current synthetic approach to them suffers from quite low overall yields (3.3% and 3.0% for 1a and 1b, respectively). In order to explore the structure-activity relationship (SAR) of 1a and 1b, synthetic approach with higher overall yield is urgently needed. In the present study, two efficient synthetic approaches to 1a and 1b were developed (approaches A and B), with CuCl-catalyzed nucleophilic aromatic substitution (SNAr) reaction of bromotriazole with sodium alkoxides and SNAr reaction of methylsulfonyltriazole with sodium alkoxides as key steps, and the conditions for important steps were fully optimized. The two synthetic approaches are characterized by dramatically higher yields, and not only valuable to the further SAR exploration of 1a and 1b but also very helpful to the synthesis of heterocycles with alkoxyl groups.
-
Key words:
- URAT1 inhibitor
- / gout
- / hyperuricemia
- / synthetic route
- / SNAr
- / CuCl catalysis
-
痛风是困扰人类千百年来的代谢性疾病之一[1~3].研究表明, 嘌呤在人体内代谢的终产物尿酸不能及时排出体外是形成高尿酸血症的重要原因, 高尿酸血症是血液中尿酸浓度高出其在血液中的溶解度(6.8 mg/dL, 404 μmmol/L)的一种疾病状态[3~4].在长期高尿酸血症的状态下, 肢体远端关节等血流较缓, 温度较低的部位, 尿酸会以其单钠盐的形式析出, 所形成的结晶会触发炎症反应而引发痛风, 主要特征是关节红肿和疼痛, 长期痛风会导致骨腐蚀和肾结石等, 甚至会威胁生命[1].随着人们生活水平的提高, 食物中摄取的嘌呤越来越多, 痛风患者的人数也在逐年增加[5].同高血压、高血脂和糖尿病等代谢病相比, 痛风一线用药品种少, 特异性不强, 副作用大, 这给痛风患者带来了极大的痛苦和不便, 市场迫切需要新的痛风治疗药物[6].尿酸转运体1 (uric acid transporter 1, URAT1) 位于肾小管上皮细胞的管腔侧, 是原尿中尿酸盐重吸收的重要参与者[7], 找到一种药物专一性抑制URAT1就能阻断尿酸盐的重吸收, 使其随尿液排出体外, 进而可达到降尿酸的目的.美国Ardea生物科技公司在临床上研究抗病毒候选药物RDEA806时, 偶然发现其有降血尿酸作用.进一步研究发现, 其体内代谢物RDEA594能特异性抑制URAT1[8], 后该公司将它命名为lesinurad, 并于2015年年底经美国食品药品监督管理局(FDA)批准在美国上市(图 1)[9].目前该公司第二代URAT1抑制剂verinurad (RDEA3170) 的开发也已进入Ⅱ期临床阶段[10].
图 1
抗病毒药物RDEA806及URAT1抑制剂lesinurad和verinurad的结构
Figure 1.
Structures of antiviral agent RDEA806 and URAT1 inhibitors lesinurad and verinurad
本实验室前期在研究URAT1抑制剂构效关系时[11], 设计了化合物1a和1b, 其中1b的体外URAT1抑制实验发现其活性与lesinurad相当, 更重要的是, 考虑到烷氧基不是三唑的常见取代基团, 因此它们在药物设计上可以作为具有新结构的药用先导化合物.但是在我们计划进一步对它们进行系统的构效关系研究时发现, 其原有合成路线反应总收率极低[Scheme 1, 3.3% (1a)和3.0% (1b)], 尤其是溴代三唑4与醇钠的亲核取代反应收率只有14% (5a)和13% (5b), 因此亟需一条收率高的合成1a和1b的路线来满足我们将1a和1b作为药用先导化合物进行进一步的构效关系研究的工作.在本文中, 经过深入研究, 我们发现了两条高效的合成路线((Scheme 2, 路线A和路线B), 它们具有收率大幅度提高的优点, 除了可以作为先导化合物1a和1b继续进行构效关系研究的合成路线外, 还可以为含烷氧基取代的其他杂环化合物的合成提供有价值的借鉴.
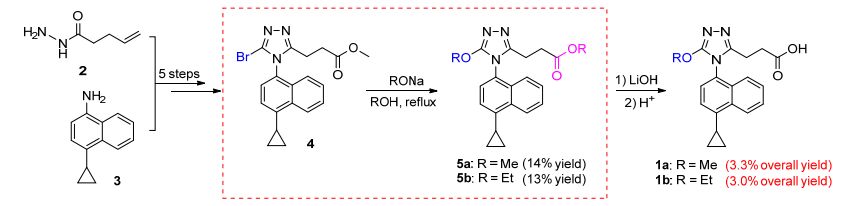 图式1
化合物1a和1b的原始合成路线
图式1.
Original synthetic route to 1a and 1b
图式1
化合物1a和1b的原始合成路线
图式1.
Original synthetic route to 1a and 1b
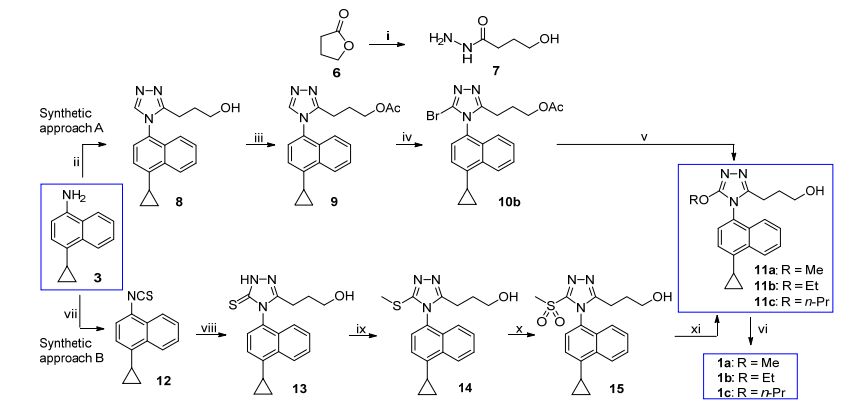 图式2
化合物1a~1c的新合成路线
图式2.
New synthetic routes to compounds 1a~1c
图式2
化合物1a~1c的新合成路线
图式2.
New synthetic routes to compounds 1a~1c
1 结果与讨论
1.1 路线设计策略分析
分析了图 2中所示醇钠对溴代三唑的芳香族亲核取代反应(SNAr), 认为1, 2, 4-三唑3位侧链部分的酯键是造成收率极低的重要原因, 因为在强碱醇钠的存在下, 酯键可能会发生酯交换和酯缩合(Claisen缩合)等副反应.因此在本研究的新路线设计中, 我们设计了在SNAr之前的路线中回避提前引入酯键, 将烷氧基引入分子后再引入酯键.考虑到卤素原子, 甲磺酰基等均可以作为离去基团, 因此设计了两条路线, 一条以卤素原子作为离去基团(Scheme 2, 路线A), 另一条以甲磺酰基作为离去基团(Scheme 2, 路线B).研究结果表明, 我们上述的分析和路线设计是正确的.
1.2 以卤素为离去基团(路线A)
如(Scheme 2中路线A所示, 化合物6在MeOH中室温下经水合肼肼解, 得到酰肼7.酰肼7与N, N-二甲基甲酰胺二甲基缩醛(DMFDMA)先反应, 而后与萘胺3反应成环, 一锅法合成得到1, 2, 4-三唑8.在4-二甲氨基吡啶(DMAP)催化下, 伯醇8在室温下的吡啶中以乙酸酐乙酰化将羟基保护, 得到乙酸酯9. 9用N-溴代丁二酰亚胺(NBS)在乙腈中室温下溴化, 得溴代三唑10b.在CuCl催化和氮气保护下, 10b与醇钠在对应的无水醇中回流反应, 得到烷氧基取代的1, 2, 4-三唑11a和11b.在NaClO/TEMPO催化下, 使用NaClO2在乙腈和磷酸缓冲液中将11a和11b中的羟基一步顺利氧化为羧基[12], 得到最终产物1a和1b, 该氧化条件对伯羟基的选择性很高, 没有干扰到被烷氧基活化的三唑环本身.使用路线A合成1a和1b的总收率为19.8%和19.9%, 相比原来路线的3.3%和3.0%有了大幅度提高.为了进一步验证路线A的通用性, 我们又利用该路线合成了R=n-Pr的化合物1c, 结果显示路线A也可以高产率地合成1c (总收率16.7%).
如Scheme 2中路线A所示, 由于担心化合物8中的羟基会干扰NBS对三唑的溴化, 因此我们将化合物8中的羟基使用乙酰基先进行了保护得到9, 然后再进行溴化; 考虑化合物9中的Br也有可能被Cl和Ⅰ代替作为离去基团, 因此我们对这些变量一并进行了优化和考察(表 1).如表 1所示, 对于卤素之间的比较, Cl, Br, Ⅰ均可使三唑9的5-位卤化, 但反应速率和收率差异很大: N-氯代丁二酰亚胺(NCS)和N-碘代丁二酰亚胺(NIS)只有在回流温度下且时间足够长时才能和原料充分反应, 收率很低(10a和10c); 而NBS在室温下即可顺利卤化, 时间较短, 收率可达到70%, 因此我们选择了Br作为离去基团.如果直接将化合物8用NBS溴化, 在室温下几乎不反应, 将温度升至35 ℃反应可以发生但是只有中等收率, 远不如羟基被乙酰基保护的9 (10d), 说明游离羟基确实可以干扰NBS对三唑的溴化, 因此在最终路线中我们选择了将8中的羟基先用乙酰基保护生成9再用NBS溴化的策略.
 表 1
不同卤化剂卤化8和9的结果比较a
Table 1.
Results for the halogenation of 8 and 9 with varying halogenating reagents
表 1
不同卤化剂卤化8和9的结果比较a
Table 1.
Results for the halogenation of 8 and 9 with varying halogenating reagents

Compd. R X T/℃ Time/h Yieldb/% 10a Ac Cl Reflux 36 26 10b Ac Br 25 24 70 10c Ac I Reflux 36 12 10d H Br 35 24 52 aReaction conditions: 8~9 (1.0 equiv.), NXS (1.1 equiv.), MeCN. bIsolated yield. 随后我们对SNAr反应中醇钠的制备方法, 反应是否需要催化剂以及底物是否需要乙酰基保护进行了仔细筛选(表 2).对于醇钠的产生方法, 我们试验了NaH/ EtOH/THF[13]和EtONa/EtOH两个体系, 前者是在THF中NaH与EtOH反应制备EtONa, 后者是将金属钠溶于无水EtOH中制得EtONa, 过量的EtOH作反应溶剂; 我们也尝试了使用CuCl作为催化剂[14]来考察是否会提高SNAr的收率.从表 2可以看出, 使用NaH与EtOH反应产生的EtONa作为反应物, 底物10b和10d中三唑环上的Br会大部分被水解, 产生大量副产物11d (表 2, Entries 1, 4), 而使用金属钠溶于EtOH后制备的EtONa/EtOH体系则水解较少(表 2, Entries 2, 3, 5, 6), 可能是NaH通常不纯(经过储存后会难以避免地与空气中水汽接触导致水解而含有一定量的NaOH), NaOH会导致10b和10d中三唑环上的Br水解, 因此使用金属钠溶于EtOH后制备的EtONa/EtOH体系是最佳选择.侧链末端无论是游离羟基还是乙酰基保护后, 对该反应收率和速率均无显著影响, 只是当用乙酰基保护时, 收率略高且副产物略少(表 2, Entry 2与Entry 5对比; Entry 3与Entry 6对比), 进一步证实了用乙酰基保护8中的羟基提高总收率的方法是合适的.值得注意的是, NaOEt少于5 equiv.时, 原料10b或10d始终不能反应完全, 增加至10 equiv.后, 反应彻底, 产物11b收率超过50%.因CuCl对醇钠的烷氧基亲核取代反应有一定催化作用[14], 本实验也考察了以CuCl为催化剂, NaOEt对溴的亲核取代反应.结果表明, CuCl可明显加快反应速率, 使反应时间缩短至三分之一, 提高产物11b收率至85%, 使副产物11d下降至10%以下(表 2, Entry 2与Entry 3对比; Entry 5与Entry 6对比).使用CuCl催化时副产物的减少可能归因于反应时间大幅度缩短, 水解程度不深.综上所述, 在SNAr反应中, 经过优化, 最终选择金属钠溶于无水EtOH制备的EtONa/EtOH体系, CuCl作为催化剂, 使用有乙酰基保护的10b作为底物.
表 2
10b和10d芳香族亲核取代反应的条件筛选a
Table 2.
Results for the screening of SNAr reaction conditions

Entry R Reagent Solvent Catalyst Time/h Yieldb/% 11b 11d 1 H NaH THF — 8 14 57 2 H NaOEt EtOH — 24 54 23 3 H NaOEt EtOH CuCl 8 84 10 4 Ac NaH THF — 8 16 41 5 Ac NaOEt EtOH — 24 54 17 6 Ac NaOEt EtOH CuCl 8 85 7 aReaction conditions: For Entries 1 and 4, 10b or 10d (1.0 equiv.), NaH (3.0 equiv.), anhydrous EtOH (1.2 equiv.), dry THF, reflux, N2; for Entries 2 and 5, Na (10 equiv.), anhydrous EtOH, 10b or 10d (1.0 equiv.), reflux, N2; for Entries 3 and 6, Na (10 equiv.), anhydrous EtOH, CuCl (0.2 equiv.), 10b or 10d (1.0 equiv.), reflux, N2.bIsolated yield. 1.3 以甲磺酰基为离去基团(路线B)
同卤素类似, 磺酰基在亲核取代反应中有时是一个很好的离去基团[15~17].我们在进行逆合成分析时设计了化合物15, 并据此设计了Scheme 2中的路线B.萘胺3在CH2Cl2中二异丙基乙基胺(DIPEA)存在下与硫光气反应, 得到异硫氰酸酯12[18]. 12先与酰肼7反应, 得到的加成产物经过NaOH处理关环, 得到三唑硫酮13[19, 20].13与碘甲烷在DMF中室温下反应得到硫醚14. 14在CH2Cl2中室温下经间氯过氧苯甲酸(mCPBA)氧化可得甲磺酰基三唑15. 15在DMF中在Cs2CO3存在下与MeOH或者EtOH发生SNAr反应, 得到11a和11b.特别需要指出的是, 由15通过SNAr反应制备11a和11b相比10b作为底物的反应, 反应条件温和, 且无水解副产物11d生成.尽管路线B比路线A多一步反应, 但是使用路线B合成1a和1b的总收率为32.5%和31.2%, 不仅相比原来路线的3.3%和3.0%有了大幅度提高, 而且相比路线A也有较大提高.同路线A一样, 我们也研究了利用路线B合成R=n-Pr的化合物1c, 来进一步验证路线B的通用性, 结果显示路线B也可以高产率地合成1c (27.6%).
在路线B中, 对化合物14到15的硫醚氧化反应条件进行了筛选(表 3).我们选择了三种常见的硫醚氧化剂, mCPBA, KMnO4和H2O2[21~23], 所有反应均在室温下操作. mCPBA氧化彻底, 后处理简便, 收率很高(Entry 1); KMnO4氧化迅速, 但体系有无机沉淀产生, 后处理时需用硅藻土助滤, 有一定量产物损失, 造成收率不高(Entry 2); H2O2氧化以钨酸钠为催化剂, 室温下几乎不反应, 增加试剂用量, 延长反应时间, 升温至60 ℃转化率依然很低(Entry 3), 可见该体系不适宜硫醚14的氧化.因此, 根据收率和操作便利性确定了以mCPBA为氧化剂的反应条件.
表 3
化合物14的氧化条件筛选
Table 3.
Screening of oxidation conditions on compound 14

Entry Reagent Solvent Time/h Yieldd/% 1a mCPBA CH2Cl2 18 93 2b KMnO4 AcOH/H2O 1 48 3c H2O2/Na2WO4 DMF 48 3 a14 (1.0 equiv.), mCPBA (2.5 equiv.), CH2Cl2, r.t.b14 (1.0 equiv.), KMnO4 (2.5 equiv.), 50% AcOH, r.t.c14 (1.0 equiv.), Na2WO4 (0.2 equiv.), H2O2(4.0 equiv.), DMF, r.t. dIsolated yield. 2 结论
本研究设计两条合成11a和11b的路线, 并对主要反应步骤进行了条件优化, 相比原来的路线具有收率大幅度提高的优点.另外, 两条路线除了可以作为药用先导化合物1a和1b继续进行构效关系研究的合成路线外, 还可以为含烷氧基取代的其他杂环化合物的合成提供有价值的借鉴.为了进一步验证路线A和B的通用性, 我们又合成了正丙基取代的三唑衍生物1c, 结果显示两条路线均可以高产率地合成1c.尽管A和B两条路线均满足合成11a~11c及其衍生物的需求, 且路线B比A多一步反应, 但是总体上来讲路线B收率相对更高, 条件更加温和.
3 实验部分
3.1 仪器与试剂
RY-2显微熔点测定仪(天津天光光学仪器有限公司), 温度未校正; Bruker AV400型核磁共振仪, DMSO-d6为溶剂, TMS内标; Agilent Q-TOF 6510型高分辨质谱仪, 电喷雾离子化技术(ESI), 直接进样法进样.各种干燥溶剂采用标准方法制备.化合物3为将市售化合物3的盐酸盐加入2 M的NaOH调至碱性, 二氯甲烷萃取, 有机相水洗, 干燥(Na2SO4)并蒸干溶剂后获得.
3.2 实验方法
3.2.1 4-羟基丁酰肼(7)的合成
γ-丁内酯(50.15 g, 583 mmol)溶于MeOH (250 mL)中, 于冰水浴冷却下缓慢加入80%水合肼(54.68 g, 874 mmol), 加毕后室温搅拌过夜.薄层色谱(TLC)检测反应完毕后, 反应混合物在旋转蒸发仪上蒸去溶剂, 将残余固体室温下加入CH2Cl2(150 mL)搅拌2 h, 抽滤收集晶体, 室温下真空干燥, 得57.04 g白色晶体7, 收率83%. m.p. 84~86.5 ℃(文献值[24]: 88~90 ℃); 1H NMR (DMSO-d6, 400 MHz) δ: 8.90 (brs, 1H), 4.19 (brs, 3H), 3.35 (t, J=6.4 Hz, 2H), 2.03 (t, J=7.6 Hz, 2H), 1.57~1.64 (m, 2H); HRMS calcd for C4H11N2O2[M+H]+ 119.0821, found 119.0820.
3.2.2 4-(4-环丙基萘-1-基)-3-(3-羟基丙基)-4H-1, 2, 4-三唑(8)的合成
化合物7 (18.93 g, 160 mmol)溶于MeCN (50 mL), 加入DMFDMA (19.10 g, 160 mmol), 50℃反应2 h, 然后将化合物3 (26.70 g, 146 mmol)溶于MeCN (30 mL)后加入到体系中, 再加入冰乙酸(20 mL), 回流12 h, TLC检测反应完成.反应混合物冷却至室温, 倾入冰水(400 mL)中, 搅拌10 min, 用CH2Cl2 (100 mL×3) 萃取.合并萃取相, 依次用饱和NaHCO3溶液(100 mL)和10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 得到的残余物经柱层析[V(MeOH)/V(EtOAc)=1/10]分离, 得19.52 g白色固体8, 收率46%. m.p. 122~124 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 8.67 (d, J=0.8 Hz, 1H), 8.57 (d, J=8.4 Hz, 1H), 7.73 (t, J=7.6 Hz, 1H), 7.64 (t, J=7.6 Hz, 1H), 7.57 (d, J=7.6 Hz, 1H), 7.40 (d, J=7.6 Hz, 1H), 7.12 (d, J=8.4 Hz, 1H), 4.38 (t, J=5.0 Hz, 1H), 3.29~3.33 (m, 2H), 2.51~2.56 (m, 1H), 2.43~2.47 (m, 2H), 1.63~1.71 (m, 2H), 1.12~1.15 (m, 2H), 0.86~0.90 (m, 1H), 0.75~0.80 (m, 1H); 13C NMR (DMSO-d6, 100 MHz) δ: 154.42, 144.92, 141.63, 133.35, 129.19, 128.24, 127.83, 127.03, 125.24, 124.93, 122.56, 121.78, 59.73, 29.83, 20.71, 12.84, 7.27, 6.81; HRMS calcd for C18H20-N3O[M+H]+ 294.1606, found 294.1610.
3.2.3 3-(3-乙酰氧基丙基)-4-(4-环丙基萘-1-基)-4H-1, 2, 4-三唑(9)的合成
化合物8 (4.26 g, 15 mmol)溶于吡啶(50 mL)中, 冰水浴冷却下搅拌, 慢慢滴加乙酸酐(20 mL), 滴加完毕后加入DMAP (0.50 g), 然后反应混合物室温下搅拌过夜, TLC检测反应完毕.将反应混合液倾入冰水(200 mL), 搅拌下加入1 mol/L HCl调pH 3~4, 再用CH2Cl2 (100 mL×3) 萃取.合并萃取相, 依次用饱和NaHCO3溶液(100 mL)和10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 得到的残余物经柱层析[V(MeOH)/ V(EtOAc)=1/20]分离, 得4.53 g无色黏稠油状物9, 收率93%.1H NMR (DMSO-d6, 400 MHz) δ: 8.70 (s, 1H), 8.56 (d, J=8.4 Hz, 1H), 7.73 (t, J=7.6 Hz, 1H), 7.63 (t, J=7.8 Hz, 1H), 7.57 (d, J=7.6 Hz, 1H), 7.40 (d, J=7.6 Hz, 1H), 7.12 (d, J=8.4 Hz, 1H), 3.93 (t, J=6.4 Hz, 2H), 2.47~2.56 (m, 3H), 1.82~1.87 (m, 2H), 1.80 (s, 3H), 1.12~1.16 (m, 2H), 0.84~0.89 (m, 1H), 0.75~0.80 (m, 1H); 13C NMR (DMSO-d6, 100 MHz) δ: 170.01, 153.64, 145.02, 141.69, 133.34, 129.11, 128.07, 127.84, 127.04, 125.24, 124.91, 122.52, 121.71, 62.62, 25.61, 20.38, 12.81, 7.22, 6.82; HRMS calcd for C20H22N3O2[M+H]+336.1712, found 336.1715.
3.2.4 卤化物10a~10d的合成
在一只100 mL圆底烧瓶中, 将化合物8 (5.50 g, 19 mmol)或者化合物9 (6.06 g, 18 mmol)溶于MeCN (50 mL)中, 室温下搅拌, 加入N-卤代丁二酰亚胺(NCS, NBS或NIS, 21 mmol), 而后在室温(9与NBS), 35 ℃ (8与NBS)或回流(NCS和NIS)下反应, 直到TLC监测反应完成(NBS: 24 h; NCS和NIS: 36 h).将反应混合物冷却至室温, 倾入冰水(200 mL)中, CH2Cl2 (100 mL×3) 萃取.合并萃取相, 依次用5% Na2S2O3溶液(100 mL), 饱和Na2CO3溶液(100 mL×2) 洗涤和10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 得到的残余物经柱层析[10a~10c: V(EtOAc)/V(n-hexane)=1/2; 10d: EtOAc]分离.
3-(3-乙酰氧基丙基)-5-氯-4-(4-环丙基萘-1-基)-4H-1, 2, 4-三唑(10a):无色黏稠油状物, 1.73 g, 收率26%. 1H NMR (DMSO-d6, 400 MHz) δ: 8.57 (d, J=8.4 Hz, 1H), 7.71~7.75 (m, 1H), 7.63~7.68 (m, 2H), 7.43 (d, J=7.6 Hz, 1H), 7.12 (d, J=8.0 Hz, 1H), 3.92 (t, J=6.2 Hz, 2H), 2.52~2.56 (m, 1H), 2.40~2.46 (m, 2H), 1.78~1.85 (m, 5H), 1.12~1.16 (m, 2H), 0.83~0.87 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 170.00, 156.38, 142.75, 141.41, 133.41, 128.79, 128.22, 127.20, 126.52, 126.26, 125.15, 122.62, 121.31, 62.48, 25.04, 21.52, 20.40, 12.81, 7.31, 7.11; HRMS calcd for C20H21ClN3O2 [M+ H]+ 370.1322, found 370.1328.
3-(3-乙酰氧基丙基)-5-溴-4-(4-环丙基萘-1-基)-4H-1, 2, 4-三唑(10b):无色黏稠油状物, 5.25 g, 收率70%. 1H NMR (DMSO-d6, 400 MHz) δ: 8.57 (d, J=8.4 Hz, 1H), 7.73 (t, J=7.6 Hz, 1H), 7.62~7.66 (m, 2H), 7.43 (d, J=8.0 Hz, 1H), 7.07 (d, J=8.4 Hz, 1H), 3.92 (t, J=6.4 Hz, 2H), 2.53~2.55 (m, 1H), 2.38~2.46 (m, 2H), 1.77~1.84 (m, 5H), 1.13~1.15 (m, 2H), 0.84~0.87 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 169.98, 156.83, 142.59, 133.41, 130.44, 128.90, 128.11, 127.14, 126.56, 125.10, 122.62, 121.46, 62.48, 25.15, 21.49, 20.38, 12.81, 7.29, 7.11; HRMS calcd for C20H2179BrN3O2[M+H]+414.0817, found 414.0811; calcd for C20H2181BrN3O2[M+H]+ 416.0797, found 416.0795.
3-(3-乙酰氧基丙基)-5-碘-4-(4-环丙基萘-1-基)-4H-1, 2, 4-三唑(10c):褐色泡沫状物, 0.98 g, 收率12%. 1H NMR (DMSO-d6, 400 MHz) δ: 8.57 (d, J=8.8 Hz, 1H), 7.70~7.74 (m, 1H), 7.60~7.64 (m, 1H), 7.56 (d, J=7.6 Hz, 1H), 7.42 (d, J=7.6 Hz, 1H), 6.98 (d, J=8.4 Hz, 1H), 3.90 (t, J=6.2 Hz, 2H), 2.51~2.56 (m, 1H), 2.43~2.47 (m, 2H), 1.75~1.82 (m, 5H), 1.12~1.17 (m, 2H), 0.84~0.87 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 170.01, 156.71, 142.32, 133.38, 129.14, 128.53, 127.94, 127.10, 126.65, 125.06, 122.58, 121.76, 106.20, 62.50, 25.41, 21.36, 20.41, 12.82, 7.24, 7.22; HRMS calcd for C20H21IN3O2 [M+H]+ 462.0678, found 462.0679.
5-溴-4-(4-环丙基萘-1-基)-3-(3-羟基丙基)-4H-1, 2, 4-三唑(10d).白色固体, 3.62 g, 收率52%. m.p. 133.5~136 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 8.57 (d, J=8.4 Hz, 1H), 7.72 (t, J=7.6 Hz, 1H), 7.62~7.66 (m, 2H), 7.42 (d, J=8.0 Hz, 1H), 7.07 (d, J=8.0 Hz, 1H), 4.32 (brs, 1H), 3.29 (t, J=6.2 Hz, 2H), 2.52~2.57 (m, 1H), 2.34~2.47 (m, 2H), 1.59~1.71 (m, 2H), 1.10~1.18 (m, 2H), 0.81~0.89 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 157.60, 142.53, 133.42, 130.25, 128.97, 128.10, 127.29, 127.13, 126.54, 125.12, 122.65, 121.51, 59.52, 29.27, 21.82, 12.84, 7.32, 7.10; HRMS calcd for C18H1979BrN3O[M+H]+372.0711, found 372.0710; calcd for C18H1981Br-N3O [M+H]+ 374.0691, found 374.0693.
3.2.5 5-烷氧基-4-(4-环丙基萘-1-基)-3-(3-羟基丙基)-4H-1, 2, 4-三唑(11a~11d)的合成[路线A]
NaH/EtOH/THF工艺:将化合物10b或10d (2.4 mmol)溶于干燥的THF (10 mL)中, 于冰水浴冷却下分批投入NaH (60%, 0.29 g, 7.2 mmol), 5 min后再滴加无水EtOH (0.13 g, 2.9 mmol), N2保护下室温搅拌30 min, 然后再回流8 h, TLC检测直到反应完成(一般8 h左右).反应结束后, 混合液冷却至室温, 小心倾入冰水(100 mL), 搅拌下用1 mol/L HCl调pH 3~4, 再以CH2Cl2 (50 mL×3) 萃取.合并萃取相, 用10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 得到的残余物经柱层析[V(MeOH)/V(EtOAc)=1/8]分离, 得11b和副产物11d.
4-(4-环丙基萘-1-基)-5-乙氧基-3-(3-羟基丙基)-4H-1, 2, 4-三唑(11b):收率14% (Table 2中的Entry 1) 和16% (Table 2中的Entry 4). m.p. 155.5~157.5 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 8.53 (d, J=8.4 Hz, 1H), 7.69 (t, J=7.6 Hz, 1H), 7.62 (t, J=7.4 Hz, 1H), 7.52 (d, J=7.6 Hz, 1H), 7.37 (d, J=7.6 Hz, 1H), 7.19 (d, J=8.0 Hz, 1H), 4.30~4.37 (m, 3H), 3.25~3.30 (m, 2H), 2.47~2.54 (m, 1H), 2.22~2.36 (m, 2H), 1.53~1.64 (m, 2H), 1.10~1.17 (m, 5H), 0.82~0.87 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 158.52, 151.91, 141.56, 133.42, 129.25, 127.63, 126.89, 126.78, 126.03, 124.95, 122.70, 121.79, 66.31, 59.68, 29.24, 21.64, 14.22, 12.79, 7.18, 6.86; HRMS calcd for C20H24N3O2[M+H]+ 338.1869, found 338.1871.
4-(4-环丙基萘-1-基)-5-羟基-3-(3-羟基丙基)-4H-1, 2, 4-三唑(11d):收率57% (Table 2中的Entry 1) 和41% (Table 2中的Entry 4). m.p. 157~159 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 11.71 (brs, 1H), 8.52 (d, J=8.4 Hz, 1H), 7.69 (t, J=7.6 Hz, 1H), 7.62 (t, J=7.4 Hz, 1H), 7.48 (d, J=7.6 Hz, 1H), 7.45 (d, J=8.0 Hz, 1H), 7.37 (d, J=7.6 Hz, 1H), 4.35 (t, J=5.2 Hz, 1H), 3.25~3.32 (m, 2H), 2.46~2.53 (m, 1H), 2.12~2.30 (m, 2H), 1.50~1.61 (m, 2H), 1.09~1.19 (m, 2H), 0.83~0.87 (m, 1H), 0.75~0.79 (m, 1H); 13C NMR (DMSO-d6, 100 MHz) δ: 154.89, 147.69, 141.07, 133.53, 129.82, 127.44, 127.26, 126.71, 126.68, 124.86, 122.73, 122.40, 59.52, 28.23, 22.31, 12.81, 7.17, 6.70; HRMS calcd for C18H20N3O2 [M+H]+ 310.1556, found 310.1550.
EtONa/EtOH工艺:盛有无水MeOH, EtOH或n-PrOH (20 mL)的50 mL圆底烧瓶用冰水浴冷却, 分批加入新切碎的金属钠(0.6 g, 24 mmol), 搅拌, 待钠完全消失后再加入化合物10b或10d (2.4 mmol)和干燥的CuCl (0.05 g, 0.48 mmol), 然后在N2保护下回流直到TLC检测反应完毕(所需时间见Table 2; 对于MeONa/MeOH, 通常需要8 h).反应完毕后, 反应混合物冷却到室温, 倾入冰水(100 mL)中, 搅拌下用1 mol/L HCl调pH 3~4, 再以CH2Cl2 (50 mL×3) 萃取.合并萃取相, 用10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 得到的残余物经柱层析[V(MeOH)/V(EtO-Ac)=1/8]分离, 得产物11a~11c.
4-(4-环丙基萘-1-基)-3-(3-羟基丙基)-5-甲氧基-4H-1, 2, 4-三唑(11a):白色固体0.68 g, 收率87%. m.p. 153.5~155 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 8.53 (d, J=8.4 Hz, 1H), 7.68~7.72 (m, 1H), 7.60~7.64 (m, 1H), 7.53 (d, J=7.6 Hz, 1H), 7.37 (d, J=7.6 Hz, 1H), 7.19 (d, J=8.4 Hz, 1H), 4.34 (t, J=5.0 Hz, 1H), 3.90 (s, 3H), 3.27 (t, J=5.8 Hz, 2H), 2.49~2.52 (m, 1H), 2.28~2.34 (m, 2H), 1.54~1.63 (m, 2H), 1.09~1.14 (m, 2H), 0.83~0.88 (m, 1H), 0.76~0.81 (m, 1H); 13C NMR (DMSO-d6, 100 MHz) δ: 159.22, 152.21, 141.67, 133.43, 129.28, 127.73, 126.90, 126.69, 126.09, 124.97, 122.70, 121.75, 59.69, 57.38, 29.29, 21.65, 12.81, 7.23, 6.80; HRMS calcd for C19H22N3O2[M+H]+ 324.1712, found 324.1714.
4-(4-环丙基萘-1-基)-3-(3-羟基丙基)-5-正丙氧基-4H-1, 2, 4-三唑(11c):白色固体0.69 g, 收率82%. m.p. 115~117 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 8.53 (d, J=8.4 Hz, 1H), 7.69 (t, J=7.4 Hz, 1H), 7.59~7.63 (m, 1H), 7.52 (d, J=7.6 Hz, 1H), 7.36 (d, J=7.6 Hz, 1H), 7.20 (d, J=8.4 Hz, 1H), 4.35 (t, J=5.0 Hz, 1H), 4.19~4.29 (m, 2H), 3.25~3.31 (m, 2H), 2.47~2.53 (m, 1H), 2.23~2.38 (m, 2H), 1.50~1.65 (m, 4H), 1.08~1.13 (m, 2H), 0.78~0.86 (m, 2H), 0.68 (t, J=7.6 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz)δ: 158.66, 151.91, 141.54, 133.41, 129.23, 127.58, 126.88, 126.80, 125.96, 124.94, 122.66, 121.81, 71.70, 59.68, 29.23, 21.65, 21.42, 12.78, 9.74, 7.14, 6.95; HRMS calcd for C21H26N3O2 [M+H]+ 352.2025, found 352.2028.
3.2.6 3-(5-烷氧基-4-(4-环丙基萘-1-基)-4H-1, 2, 4-三唑-3-基)丙酸(1a~1c)的合成
在100 mL圆底烧瓶中依次加入11a~11c (2.3 mmol), 2, 2, 6, 6-四甲基哌啶氧化物(TEMPO, 0.07 g, 0.46 mmol), MeCN (20 mL), 0.67 mol/L的NaH2PO4溶液(15 mL)和亚氯酸钠(80%, 0.52 g, 4.6 mmol), 而后在35 ℃下搅拌.取市售次氯酸钠溶液(质量分数21%, 0.41 g, 1.2 mmol)溶于水(10 mL)中, 慢慢滴加到体系中, 保持35 ℃反应过夜, TLC显示反应完毕.反应混合物冷却至室温, 倾入冰水(100 mL)中, 1 mol/L HCl调pH 1~2, 再以CH2Cl2 (100 mL×3) 萃取.合并萃取相, 依次用1% Na2SO3溶液(100 mL)和水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 残余物经纯EtOAc柱层析, 得到化合物1a~1c.
3-(4-(4-环丙基萘-1-基)-5-甲氧基-4H-1, 2, 4-三唑-3-基)丙酸(1a).白色固体, 0.59 g, 收率76%. m.p. 168 ℃(dec.)[文献值[11] 167 ℃ (dec.)].
3-[4-(4-环丙基萘-1-基)-5-乙氧基-4H-1, 2, 4-三唑-3-基]丙酸(1b).白色固体, 0.63 g, 收率78%. m.p. 155~156 ℃(文献值[11] 154.5~155.5℃).
3-[4-(4-环丙基萘-1-基)-5-正丙氧基-4H-1, 2, 4-三唑-3-基]丙酸(1c):无色粘稠油状物, 0.57 g, 收率68%. 1H NMR (DMSO-d6, 400 MHz) δ: 12.14 (brs, 1H), 8.53 (d, J=8.4 Hz, 1H), 7.68~7.72 (m, 1H), 7.59~7.63 (m, 1H), 7.52 (d, J=7.6 Hz, 1H), 7.38 (d, J=7.6 Hz, 1H), 7.24 (d, J=8.4 Hz, 1H), 4.20~4.30 (m, 2H), 2.47~2.56 (m, 4H), 2.32~2.41 (m, 1H), 1.51~1.60 (m, 2H), 1.09~1.16 (m, 2H), 0.81~0.85 (m, 2H), 0.69 (t, J=7.6 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz) δ: 173.05, 158.82, 150.97, 141.69, 133.51, 129.22, 127.56, 126.91, 126.67, 125.99, 124.94, 122.73, 122.00, 71.84, 29.84, 21.47, 20.40, 12.83, 9.74, 7.04, 7.01; HRMS calcd for C21H24N3O3[M+H]+ 366.1818, found 366.1823.
3.2.7 (4-环丙基萘-1-基)异硫氰酸酯(12)的合成
化合物3 (9.52 g, 52 mmol)溶于CH2Cl2 (100 mL)中, 加入DIPEA (13.43 g, 104 mmol), 冰水浴冷却下搅拌, 缓慢滴加硫光气(CSCl2, 6.57 g, 57 mmol).滴加完毕后, 撤去冰水浴, 在室温下继续搅拌30 min. TLC检测发现反应完成.反应混合物小心倾倒至搅拌的冰水(400 mL)中, 继续搅拌1 min, CH2Cl2 (100 mL×3) 萃取, 合并萃取相, 依次用0.5 mol/L HCl (200 mL)和10%食盐水(200 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 残余物经正己烷柱层析, 得10.18 g无色油状液体12, 收率87%. 1H NMR (DMSO-d6, 400 MHz) δ: 8.47 (d, J=8.0 Hz, 1H), 8.01 (dd, J=1.8, 5.8 Hz, 1H), 7.69~7.75 (m, 2H), 7.55 (dd, J=2.8, 8.0 Hz, 1H), 7.23 (dd, J=1.6, 7.6 Hz, 1H), 2.40~2.45 (m, 1H), 1.05~1.09 (m, 2H), 0.72~0.75 (m, 2H).该1H NMR数据与文献值[18]一致.
3.2.8 4-(4-环丙基萘-1-基)-5-(3-羟基丙基)-2, 3-二氢-4H-1, 2, 4-三唑啉-3-硫酮(13)的合成
化合物7 (2.36 g, 20 mmol)溶于EtOH (150 mL), 搅拌下加入化合物12 (4.50 g, 20 mmol), 升温回流过夜, 有白色不溶物析出.待体系冷却至室温后, 抽滤除去不溶物, 收集滤液, 并在旋转蒸发仪上蒸去溶剂, 向残余物中加入1 mol/L NaOH溶液(50 mL), 45 ℃加热, 直到TLC检测发现反应完成(一般1.5 h).反应混合物冷却后, 倾入冰水(200 mL)中, 加1 mol/L HCl调至pH 5~6, CH2Cl2 (100 mL×3) 萃取, 合并萃取相, 用10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 残余物经柱层析[V(EtOAc)/V(n-hexane)=1/2]分离, 得到4.78 g白色固体13, 收率74%. m.p. 221~223 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 13.79 (brs, 1H), 8.53 (d, J=8.4 Hz, 1H), 7.68 (t, J=7.8 Hz, 1H), 7.59 (t, J=7.4 Hz, 1H), 7.48 (d, J=7.6 Hz, 1H), 7.39 (d, J=7.6 Hz, 1H), 7.24 (d, J=8.4 Hz, 1H), 4.35 (brs, 1H), 3.26 (t, J=6.0 Hz, 2H), 2.51~2.53 (m, 1H), 2.25~2.33 (m, 1H), 2.15~2.22 (m, 1H), 1.54~1.63 (m, 2H), 1.10~1.16 (m, 2H), 0.83~0.87 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 168.35, 152.94, 141.59, 133.52, 129.19, 128.20, 127.31, 127.06, 126.70, 124.90, 122.69, 122.42, 59.35, 28.45, 21.97, 12.82, 7.25, 6.88; HRMS calcd for C18H20N3OS[M+H]+ 326.1327, found 326.1325.
3.2.9 4-(4-环丙基萘-1-基)-5-甲硫基-3-(3-羟基丙基)-4H-1, 2, 4-三唑(14)的合成
向100 mL圆底烧瓶中依次加入化合物13 (4.18 g, 13 mmol), DMF (50 mL), K2CO3 (1.95 g, 14 mmol)和碘甲烷(2.01 g, 14 mmol), 室温下搅拌直到TLC检测反应完毕(一般2 h).将反应混合液倾入冰水(100 mL), 用1M HCl调至pH 5~6, 再用CH2Cl2 (100 mL×3) 萃取.合并萃取相, 以10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 残余固体在V(EtOAc)/V(n-hexane)=1/3中结晶, 得到4.12 g白色固体14, 收率94%. m.p. 183.5~185 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 8.55 (d, J=8.8 Hz, 1H), 7.71 (t, J=7.6 Hz, 1H), 7.61 (t, J=7.4 Hz, 1H), 7.55 (d, J=7.6 Hz, 1H), 7.39 (d, J=7.6 Hz, 1H), 7.07 (d, J=8.4 Hz, 1H), 4.35 (t, J=5.0 Hz, 1H), 3.26~3.31 (m, 2H), 2.51~2.55 (m, 1H), 2.49 (s, 3H), 2.34~2.38 (m, 2H), 1.59~1.67 (m, 2H), 1.12~1.14 (m, 2H), 0.83~0.86 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 156.36, 151.68, 142.18, 133.48, 128.96, 127.84, 127.22, 127.03, 126.22, 125.05, 122.65, 121.71, 59.64, 29.58, 21.34, 14.42, 12.81, 7.24, 7.09; HRMS calcd for C19H22N3OS [M+H]+ 340.1484, found 340.1489.
3.2.10 4-(4-环丙基萘-1-基)-3-(3-羟基丙基)-5-甲磺酰基-4H-1, 2, 4-三唑(15)的合成
使用mCPBA氧化的工艺:向50 mL圆底烧瓶中加入化合物14 (1.00 g, 2.9 mmol), 以CH2Cl2 (20 mL)溶解, 搅拌下加入mCPBA (80%, 1.60 g, 7.4 mmol), 然后室温下反应直到TLC检测反应完成(一般18 h).反应混合物倾入冰水(200 mL)中, 用CH2Cl2 (100 mL×3) 萃取.合并萃取相, 依次用饱和NaHCO3溶液(100 mL×2) 和10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 析出的固体在V(EtOAc)/V(n-hexane)=1/3中结晶, 得到1.01 g白色固体15, 收率93%. m.p. 174.5~176.5 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 8.55 (d, J=8.8 Hz, 1H), 7.68~7.72 (m, 2H), 7.61 (t, J=7.6 Hz, 1H), 7.39 (d, J=7.6 Hz, 1H), 7.10 (d, J=7.6 Hz, 1H), 4.38 (t, J=5.2 Hz, 1H), 3.35 (s, 3H), 3.28~3.32 (m, 2H), 2.51~2.58 (m, 1H), 2.35 (t, J=7.6 Hz, 2H), 1.69~1.76 (m, 2H), 1.12~1.18 (m, 2H), 0.84~0.87 (m, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 158.70, 152.96, 142.58, 133.18, 129.00, 127.91, 127.02, 126.68, 126.35, 124.91, 122.22, 121.76, 59.46, 43.38, 29.24, 20.98, 12.80, 7.32, 7.26; HRMS calcd for C19H22N3O3S [M+H]+ 372.1382, found 372.1384.
使用KMnO4氧化的工艺:向50 mL圆底烧瓶中加入化合物14 (1.00 g, 2.9 mmol)、50%乙酸(20 mL)和KMnO4 (0.93 g, 5.9 mmol), 室温反应直到TLC检测反应(一般1 h).反应混合物用硅藻土过滤, 滤液倒入冰水(200 mL)中, 用CH2Cl2 (100 mL×3) 萃取.合并萃取相, 依次用饱和NaHCO3溶液(100 mL)和10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 析出的固体在V(EtOAc)/V(n-hexane)=1/3中结晶, 得到0.52 g白色固体15, 收率48%.
使用H2O2氧化的工艺:向50 mL圆底烧瓶中加入化合物14 (1.00 g, 2.9 mmol), 钨酸钠(0.19 g, 0.59 mmol)和DMF (20 mL), 40 ℃搅拌下滴加入H2O2 (30%, 1.34 g, 12 mmol), 升温至60 ℃反应48 h, TLC显示体系仍有大量原料存在, 延长时间原料未见减少.将反应混合物冷却至室温, 倾入冰水(200 mL)中, CH2Cl2 (100 mL×3) 萃取.合并萃取相, 以10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 得到的残余物经柱层析[V(EtOAc)/V(n-hexane)=1/2]分离, 得到0.03 g白色固体15, 收率3%.
3.2.11 5-烷氧基-4-(4-环丙基萘-1-基)-3-(3-羟基丙基)-4H-1, 2, 4-三唑(11a~11c)的合成[路线B]
化合物15 (0.80g, 2.2 mmol)溶于DMF (15 mL)中, 加入碳酸铯(2.11 g, 6.5 mmol), MeOH, EtOH或n-PrOH (5 mL), 80 ℃下反应24 h, TLC监测反应完成.将反应混合物冷却至室温, 倾入冰水(200 mL), 搅拌下用1 mol/L HCl调pH 3~4, 再以CH2Cl2 (100 mL×3) 萃取.合并萃取相, 用10%食盐水(100 mL)洗涤, 无水Na2SO4干燥, 蒸去溶剂, 得到的残余物经柱层析[V(MeOH)/ V(EtOAc)=1/8]分离, 得产物11a (0.53 g, 收率76%), 11b (0.52 g, 收率71%)和11c (0.56 g, 收率72%).
辅助材料(Supporting Information) 合成的所有新化合物的1H NMR和13C NMR图谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
Richette, P.; Bardin T. Lancet 2010, 375, 318. doi: 10.1016/S0140-6736(09)60883-7
-
[2]
Pillinger, M. H.; Rosenthal, P.; Abeles, A. M. Bull. NYU Hosp. Jt. Dis. 2007, 65, 215.
-
[3]
Punzi, L.; Scanu, A.; Ramonda, R.; Oliviero, F. Autoimmun. Rev. 2012, 12, 66. doi: 10.1016/j.autrev.2012.07.024
-
[4]
Choi, H. K.; Mount, D. B.; Reginato, A. M. Ann. Intern. Med. 2005, 143, 499. doi: 10.7326/0003-4819-143-7-200510040-00009
-
[5]
Miao, Z.; Li, C.; Chen, Y.; Zhao, S.; Wang, Y.; Wang, Z.; Chen, X.; Xu, F.; Wang, F.; Sun, R.; Hu, J.; Song, W.; Yan, S.; Wang, C. J. Rheumatol. 2008, 35, 1859.
-
[6]
Dubchak, N.; Falasca, G. F. Int. J. Nephrol. Renovasc. Dis. 2010, 3, 145.
-
[7]
Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S. H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; Matsuo, H.; Kikuchi, Y.; Oda, T.; Ichida, K.; Hosoya, T.; Shimokata, K.; Niwa, T.; Kanai, Y.; Endou, H. Nature 2002, 417, 447.
-
[8]
Adams, J. U. Nat. Biotechnol. 2009, 27, 309. doi: 10.1038/nbt0409-309
-
[9]
Hoy, S. M. Drugs 2016, 76, 509. doi: 10.1007/s40265-016-0550-y
-
[10]
Miner, J. N.; Tan, P. Ann. Rheum. Dis. 2013, 71, 446.
-
[11]
Tian, H.; Liu, W.; Zhou, Z.; Shang, Q.; Liu, Y.; Xie, Y.; Liu, C.; Xu, W.; Tang, L.; Wang, J.; Zhao, G. Molecules 2016, 21, 1543. doi: 10.3390/molecules21111543
-
[12]
Zhao, M.; Li, J.; Mano, E.; Song, Z.; Tschaen, D. M.; Grabowski, E, J. J.; Reider, P. J. J. Org. Chem. 1999, 64, 2564. doi: 10.1021/jo982143y
-
[13]
Hirata, K.; Kotoku, M.; Seki, N.; Maeba, T.; Maeda, K.; Hirashima, S.; Sakai, T.; Obika, S.; Hori, A.; Hase, Y.; Yamaguchi, T.; Katsuda, Y.; Hata, T.; Miyagawa, N.; Arita, K.; Nomura, Y.; Asahina, K.; Aratsu, Y.; Kamada, M.; Adachi, T.; Noguchi, M.; Doi, S.; Crowe, P.; Bradley, E.; Steensma, R.; Tao, H.; Fenn, M.; Babine, R.; Li, X.; Thacher, S.; Hashimoto, H.; Shiozaki, M. ACS Med. Chem. Lett. 2016, 7, 23. doi: 10.1021/acsmedchemlett.5b00253
-
[14]
Ragan, J. A.; Makowski, T. W.; Castaldi, M. J.; Hill, P. D. Synthesis 1998, 1599.
-
[15]
Bonanomi, G.; Braggio, S.; Capelli, A. M.; Checchia, A.; Di Fabio, R.; Marchioro, C.; Tarsi, L.; Tedesco, G.; Terreni, S.; Worby, A.; Heibreder, C.; Micheli, F. ChemMedChem 2010, 5, 705. doi: 10.1002/cmdc.201000026
-
[16]
谢明胜, 唐浩鑫, 张一铭, 郭真, 郭海明, 渠桂荣, 有机化学, 2015, 35, 2589. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345185.shtmlXie, M.; Tang, H.; Zhang, Y.; Guo, Z.; Guo, H.; Qu, G. Chin. J. Org. Chem. 2015, 35, 2589(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345185.shtml
-
[17]
傅颖, 赵兴玲, 侯博, 有机化学, 2016, 36, 1184. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345409.shtmlFu, Y.; Zhao, X.; Hou, B. Chin. J. Org. Chem. 2016, 36, 1184(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345409.shtml
-
[18]
Cai, W.; Liu, W.; Xie, Y.; Wu, J.; Liu, Y.; Liu, C.; Xu, W.; Tang, L.; Wang, J.; Zhao, G. Chem. Res. Chin. Univ. 2017, 33, 49. doi: 10.1007/s40242-017-6351-3
-
[19]
Reddy, T. R.; Li, C.; Guo, X.; Fischer, P. M.; Dekker, L. V. Bioorg. Med. Chem. 2014, 22, 5378. doi: 10.1016/j.bmc.2014.07.043
-
[20]
Colanceska-Ragenovic, K.; Dimova, V.; Kakurinov, V.; Gabor, D. M. J. Heterocycl. Chem. 2003, 40, 905. doi: 10.1002/jhet.v40:5
-
[21]
Kane, J. M.; Staeger, M. A.; Dalton, C. R.; Miller, F. P.; Dudley, M. W.; Ogden, A. M.; Kehne, J. H.; Ketteler, H. J.; McCloskey, T. C.; Senyah, Y.; Chmielewski, P. A.; Miller, J. A. J Med Chem. 1994, 37, 125. doi: 10.1021/jm00027a015
-
[22]
Ström, P.; Malmquist, J. J. Labelled Compd. Radiopharm. 2008, 51, 419. doi: 10.1002/jlcr.v51:13
-
[23]
Liu, M.; Shi, D. J. Heterocycl. Chem. 2014, 51, E335. doi: 10.1002/jhet.v51.S1
-
[24]
Huisgen, R.; Möbius, L.; Szeimies, G. Chem. Ber. 1965, 98, 1138. doi: 10.1002/(ISSN)1099-0682
-
[1]
-

图 1 抗病毒药物RDEA806及URAT1抑制剂lesinurad和verinurad的结构
Figure 1 Structures of antiviral agent RDEA806 and URAT1 inhibitors lesinurad and verinurad

图式2 化合物1a~1c的新合成路线
Scheme 2 New synthetic routes to compounds 1a~1c
Reagents and conditions: (ⅰ) 80% N2H4·H2O, MeOH, r.t., yield 83%; (ⅱ) (a) DMFDMA, 7, MeCN, 50 ℃; (b) 3, AcOH, reflux, yield 46%; (ⅲ) Ac2O, DMAP, pyridine, 0 ℃-r.t., yield 93%; (ⅳ) NBS, MeCN, r.t., yield 70%; (ⅴ) MeONa/MeOH (11a), EtONa/EtOH (11b) or n-PrONa/n-PrOH (11c), CuCl, reflux, N2, yield 87% (11a), 84% (11b), 82% (11c); (ⅵ) NaClO2, TEMPO, NaClO, MeCN, phosphate buffer, 35 ℃, yield 76% (1a), 78% (1b), 68% (1c); (ⅶ) CSCl2, DIPEA, CH2Cl2, 0 ℃-r.t., yield, 87%; (ⅷ) (a) 7, EtOH, reflux; (b) 1 mol/L NaOH, 45 ℃, yield 74%; (ⅸ) MeI, K2CO3, DMF, r.t., yield 94%; (ⅹ) mCPBA, CH2Cl2, r.t., yield 93%; (xi) Cs2CO3, DMF, MeOH (11a), EtOH (11b), or n-PrOH (11c), 80 ℃, yield 76% (11a), 71% (11b), 72% (11c).
表 1 不同卤化剂卤化8和9的结果比较a
Table 1. Results for the halogenation of 8 and 9 with varying halogenating reagents
Compd. R X T/℃ Time/h Yieldb/% 10a Ac Cl Reflux 36 26 10b Ac Br 25 24 70 10c Ac I Reflux 36 12 10d H Br 35 24 52 aReaction conditions: 8~9 (1.0 equiv.), NXS (1.1 equiv.), MeCN. bIsolated yield.  下载: 导出CSV
下载: 导出CSV
表 2 10b和10d芳香族亲核取代反应的条件筛选a
Table 2. Results for the screening of SNAr reaction conditions
Entry R Reagent Solvent Catalyst Time/h Yieldb/% 11b 11d 1 H NaH THF — 8 14 57 2 H NaOEt EtOH — 24 54 23 3 H NaOEt EtOH CuCl 8 84 10 4 Ac NaH THF — 8 16 41 5 Ac NaOEt EtOH — 24 54 17 6 Ac NaOEt EtOH CuCl 8 85 7 aReaction conditions: For Entries 1 and 4, 10b or 10d (1.0 equiv.), NaH (3.0 equiv.), anhydrous EtOH (1.2 equiv.), dry THF, reflux, N2; for Entries 2 and 5, Na (10 equiv.), anhydrous EtOH, 10b or 10d (1.0 equiv.), reflux, N2; for Entries 3 and 6, Na (10 equiv.), anhydrous EtOH, CuCl (0.2 equiv.), 10b or 10d (1.0 equiv.), reflux, N2.bIsolated yield.
下载: 导出CSV
表 3 化合物14的氧化条件筛选
Table 3. Screening of oxidation conditions on compound 14
Entry Reagent Solvent Time/h Yieldd/% 1a mCPBA CH2Cl2 18 93 2b KMnO4 AcOH/H2O 1 48 3c H2O2/Na2WO4 DMF 48 3 a14 (1.0 equiv.), mCPBA (2.5 equiv.), CH2Cl2, r.t.b14 (1.0 equiv.), KMnO4 (2.5 equiv.), 50% AcOH, r.t.c14 (1.0 equiv.), Na2WO4 (0.2 equiv.), H2O2(4.0 equiv.), DMF, r.t. dIsolated yield.
下载: 导出CSV
-




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 1955
- HTML全文浏览量: 261
 下载:
下载:
