Received Date:
01 January 2019 Available Online:
15 May 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 51605034)
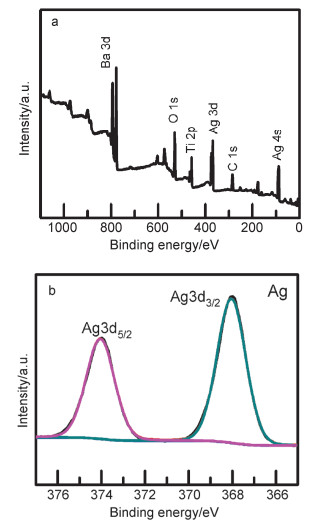
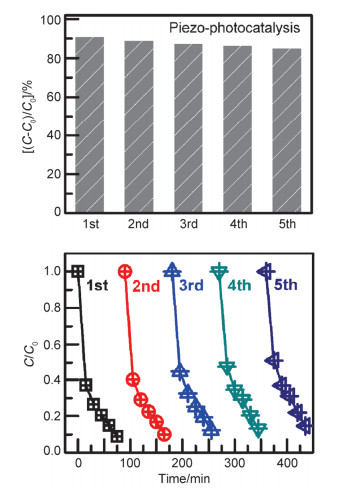
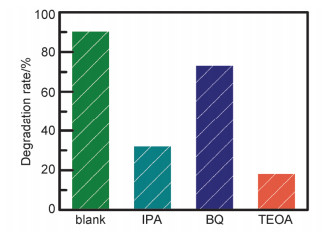
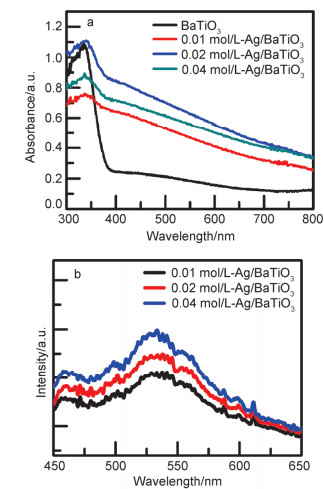
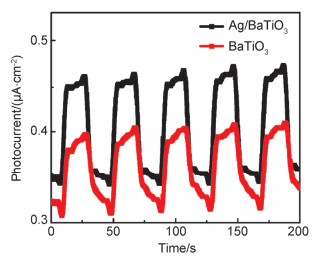
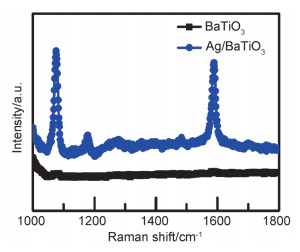
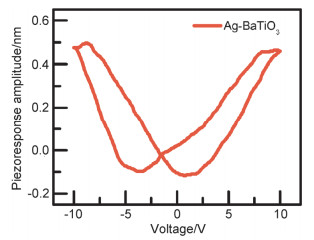
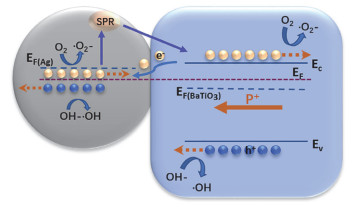
Abstract:x mol/L-Ag/BaTiO3 (x=0.01, 0.02, 0.04, where x is concentration of Ag) plasmonic photocatalysts were fabricated by precipitating Au nanoparticles on BaTiO3 nano-piezoelectric through a photochemical reducing approach. The plasmonic piezo-photocatalytic composite material can simultaneously solve the problems of low photocatalytic efficiency and narrow light absorption range in the photocatalysis process. BaTiO3 nano-piezoelectric were synthesized by a hydrothermal synthesis, Ag nanoparticles were deposited on the surface of BaTiO3 powder using a photoreduction reaction. Subsequently, the effects of microtopography, optical properties and degradation of dye were discussed by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-Ray photoelectron spectroscopy (XPS), UV-visible absorption spectra, photocurrent, photoelectrocatalytic, etc. The mechanism of piezoelectric photocatalysis and the effect of the concentration of ionic particles on the properties of the composite photocatalyst were investigated. The intensity and excitation mode of localized surface plasmon resonance (LSPR) vary on account of the different densities of nanoparticles, the 0.02 mol/L Ag-BaTiO3 showed an excellent photocatalytic performance for degrading 91% RhB in 75 min under full-spectrum light irradiation with ultrasonic excitation which can produce piezoelectric charges on the surfaces of the BaTiO3 nanocubes, and the degradation efficiency is increased by 21%. The effects of hybrid structure piezoelectric potential in nano-piezoelectric has been confirmed to express a great influence on surface plasmon resonance photocatalytic activity. The improvement of catalytic performance is due to the synergistic effect of piezoelectric effect and surface plasmon resonance effect. The LSPR of Ag nanoparticles that uniformly decorated on the surface of BaTiO3 nano-piezoelectric, widen the range of light absorption from ultraviolet to visible light. With introducing ultrasonic excitation to renew the piezoelectric charges on the surfaces of the BaTiO3 nanocubes, the piezoelectric field originated from the deformation of BaTiO3 nanotubes can further enhance the separation of photo-carriers induced by the localized surface plasmon resonance (LSPR), and promote the generation of hydroxyl radicals with strong oxidizing ability and accelerate the degradation of organic dyes. This work based on the piezotronic effect of the BaTiO3 nanocubes, assisting the surface plasmon resonance in photocatalysis improved the degradation efficiency of Rh B significantly. In addition, this discovery could be extended to other material systems to provide an effective technology for environment purification.
Figure 2.
(a) SEM image of 0.01 mol/L-Ag/BaTiO3, (b) SEM image of 0.02 mol/L-Ag/BaTiO3, (c) SEM image of 0.04 mol/L-Ag/BaTiO3, (d) TEM image of 0.02 mol/L-Ag/BaTiO3, (e) and (f) HRTEM image of 0.02 mol/L-Ag/BaTiO3
Figure 4.
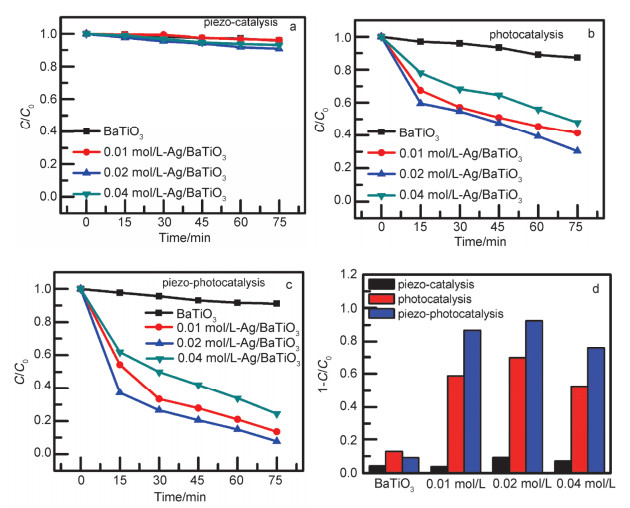
Degradation of Rh B under (a) piezoelectric excitation, (b) full-spectrum light irradiation, (c) both piezoelectric and full-spectrum light irradiation, (d) piezo-catalytic, piezo-photocatalytic, and photocatalytic degradation of RhB in the presence of x mol/L-Ag/BaTiO3 and BaTiO3 for 75 min
Chen, J.; Wu, J.; Wu, P.; Tsai, D. J. Phys. Chem. C2010, 115, 210.
[30]
Yoo, J.; Altomare, M.; Mokhtar, M.; Alshehri, A.; Al-Thabaiti, S. A.; Mazare, A.; Schmuki, P. J. Phys. Chem. C2016, 120, 15884. doi: 10.1021/acs.jpcc.5b12050
Figure 2
(a) SEM image of 0.01 mol/L-Ag/BaTiO3, (b) SEM image of 0.02 mol/L-Ag/BaTiO3, (c) SEM image of 0.04 mol/L-Ag/BaTiO3, (d) TEM image of 0.02 mol/L-Ag/BaTiO3, (e) and (f) HRTEM image of 0.02 mol/L-Ag/BaTiO3
Figure 4
Degradation of Rh B under (a) piezoelectric excitation, (b) full-spectrum light irradiation, (c) both piezoelectric and full-spectrum light irradiation, (d) piezo-catalytic, piezo-photocatalytic, and photocatalytic degradation of RhB in the presence of x mol/L-Ag/BaTiO3 and BaTiO3 for 75 min


 下载:
下载:


 下载:
下载:












