 图 1
金属氧化物催化羧酸酮基化反应机理(β-酮酸机理)示意图
Figure 1.
β-ketoacid based mechanism for ketonization of carboxylic acid over metal oxide
图 1
金属氧化物催化羧酸酮基化反应机理(β-酮酸机理)示意图
Figure 1.
β-ketoacid based mechanism for ketonization of carboxylic acid over metal oxide
引用本文:
丁爽, 葛庆峰, 祝新利. 金属氧化物催化生物质衍生羧酸酮基化研究进展[J]. 化学学报,
2017, 75(5): 439-447.
doi:
10.6023/A17020061

Citation: Ding Shuang, Ge Qingfeng, Zhu Xinli. Research Progress in Ketonization of Biomass-derived Carboxylic Acids over Metal Oxides[J]. Acta Chimica Sinica, 2017, 75(5): 439-447. doi: 10.6023/A17020061
Citation: Ding Shuang, Ge Qingfeng, Zhu Xinli. Research Progress in Ketonization of Biomass-derived Carboxylic Acids over Metal Oxides[J]. Acta Chimica Sinica, 2017, 75(5): 439-447. doi: 10.6023/A17020061
金属氧化物催化生物质衍生羧酸酮基化研究进展
摘要:
从可再生的木质纤维素生物质制备液体燃料受到越来越多的关注.有机羧酸是生物质解聚生物油的重要成分,使得生物油具有酸性、腐蚀性和不稳定性.因而,羧酸的去除十分关键.酮基化反应将两分子羧酸转化为酮、二氧化碳和水,不使用氢气的情况下高效脱氧且增加碳链长度.此外,生成的酮为重要化学品.目前酮基化反应的机理和活性位的研究还存在争论.因酮基化反应过程生成的中间产物不同(如β-酮酸、酮烯、羧化物、酰基碳正离子等),研究者们提出了不同的反应机理,如β-酮酸机理和酮烯机理.酮基化反应属于结构敏感性反应,因此金属氧化物表面结构的不同会导致酮基化反应活性不同.酸碱位协同作用在酮基化反应过程中是必不可少的,同时氧空位可以提高酮基化反应的活性.本综述重点从酮基化反应机理、金属氧化物的表面结构、酸碱性及氧化还原性方面对酮基化反应进行了评述,并对其进行了展望.
English
Research Progress in Ketonization of Biomass-derived Carboxylic Acids over Metal Oxides
Abstract:
With the increasing needs for transportable fuels and the growing concerns on environmental pollution, significant attention has been paid to the conversion of renewable lignocellulosic biomass to liquid fuels. As a major component of bio-oil from biomass depolymerization, organic carboxylic acids make the bio-oil acidic, corrosive and unstable, which are harmful for storage, transportation, and upgrading of bio-oil. Therefore, the removal of carboxylic acids is very important. Ketonization reaction, also called ketonic decarboxylation, converts two moles carboxylic acids to ketone (symmetrical or asymmetrical ketones), carbon dioxide and water, which removes oxygen efficiently and increases the carbon chain length without using hydrogen. In addition, ketones are important chemicals and have been widely used in chemical industry as organic solvent. The mechanism and active site for ketonization are still under debate. Various mechanisms have been proposed for the ketonization, based on different reaction intermediates evolved (i.e., β-keto-acids, ketene, carboxylates and acyl carbonium ions). Ketonization reaction is a surface-structure-sensitive reaction, thus reaction activity depends on surface-structure of the metal oxides (such as crystal surfaces and particle size). The concerted function of oxygen anions (Brønsted bases) and unsaturated metal cations (Lewis acids) is crucial for ketonization. The amphoteric oxides show better catalytic activity than pure acidic or basic oxides. Oxygen vacancy formed on the surface of metal oxides is a key factor for high ketonization activity, which can stabilize the reaction product and reduce the activation energy. This paper reviews the progress in ketonization from the aspects of reaction mechanism, and the effects of surface structure, acidity and basicity, and reducibility of metal oxides on ketonization. The β-keto-acids based mechanism and ketene based mechanism will be discussed in detail to understand how does the C—C coupling happen and the fundamental role of α-H. Finally, the importance of surface structure and properties of metal oxides on the carboxylic acids ketonization reaction is explained.
-
Key words:
- carboxylic acid
- / ketonization
- / metal oxides
- / surface-structure and property
- / reaction mechanism
-
1 引言
随着经济的快速发展和人口的不断增长, 对能源的需求越来越多.煤、石油、天然气这些非可再生能源的日益消耗, 不仅造成了环境污染, 全球气候变暖等问题, 而且也带来了能源危机等一系列严重后果.生物质能源是一种可再生的清洁资源, 不仅储量丰富而且分布范围广泛.生物质资源的开发和利用已经得到越来越多的关注[1~6].不同的化学方法可以将生物质转化成含C、H、O的有机化合物(如醇、醛、羧酸、酚、芳香烃类等小分子含氧有机化合物), 最常用的方法是快速热解和水解反应[7].这些小分子有机化合物还可以通过进一步转化生成汽油、柴油和其他化学品[1].生物油主要是由木质素转化生成的, 羧酸是生物油中的重要组成部分. Sedran等[8]对生物油中的组成进行了测定, 羧酸的含量高达17%.羧酸具有不稳定性, 容易发生聚合生成大分子物质, 且具有腐蚀性能损坏反应设备及管路, 所以羧酸的进一步转化是首先要解决的问题.羧酸可以通过选择性加氢生成醇或醛, 还可以通过酮基化反应生成酮.
酮基化反应转化羧酸具有如下优点.第一, 反应绿色无污染, 并且不需要溶剂和其他试剂; 第二, 反应不需要氢气就可以将有机物中的氧移除; 第三, 反应所需要的催化剂一般都是低成本的; 第四, 在转化掉高活性的羧基的同时又增长了有机物碳链的长度, 生成了更稳定的化合物.生成的酮类化合物可以继续进行羟醛缩合反应, 进一步转化成其他有机大分子化合物.
酮基化反应(也称脱羧酮基化反应)是将两分子的羧酸脱去一个羰基生成酮、二氧化碳和水, 如反应式(1) 所示.相同的两个羧酸分子(R1=R2)进行酮基化反应, 称为自身酮基化; 两个不同的羧酸分子(R1≠R2)进行反应则称为交叉酮基化[9~15]. Sato等[10]报道了丙酸与其它羧酸反应, 生成物中包括一种不对称的酮和两种对称的酮, 生成物的组成遵循二项式分布.
早在1858年, Friedel[16]首次报道了酮基化反应, 通过醋酸钙裂解来制备丙酮.直到第一次世界大战前, 该过程被用来工业上制备丙酮.经过多年的研究, 研究者逐步开发出金属氧化物、沸石分子筛等催化剂, 直接催化羧酸酮基化反应生成酮.目前工业上利用氧化锆、氧化铝负载型催化剂催化羧酸酮基化反应, 制成的酮化学品是甲基异丁基酮(MIPK).在高温催化剂作用下, 异丁酸和乙酸进行交叉酮基化反应生成MIPK.德国康得阿维斯塔公司通过异丁酸与乙酸酮基化反应生成MIPK, 每年生产能力达1500 t[17].国内的吉化公司化肥厂通过氧化锆催化异丁酸与乙酸反应制备酮的年生产能力达到250 t[17], 同时北京化工研究院自1998年自行开发了异丁酸与乙酸合成MIPK技术, 建立了每年300 t的小规模装置.虽然金属氧化物催化羧酸酮基化反应在工业上有成功的应用, 但仍存在着反应温度高、催化剂活性低、积炭造成的稳定性差等问题.这些问题的解决需要从机理上更好地认知酮基化反应.
针对羧酸酮基化反应, 在理论和实验研究过程中, 研究者们分别从α-H的作用[10, 18]、取代基空间位阻[9, 10]、羧酸碳链长度[10, 19]、中间产物[20, 21]、催化剂种类[22~28]、反应机理[29~31]及动力学[32~34]上做了不同程度探索.但对于催化剂活性位、反应机理、α-H的作用仍存在着一定的争论.与此同时, 也有研究者对酮基化反应进行过综述性报道.早在1994年, Rajadurai等[35]就过渡金属氧化物催化羧酸反应路径进行了系统的整理, 通过对中间物种类和动力学数据分析提出了羧酸酮基化反应的酮烯机理. Renz等[36]也从机理方面综述了羧酸酮基化反应, 文中指出酮基化反应中弱碱可以提高催化活性.宁朋歌等[37]从催化剂、反应物、反应温度和反应机理方面介绍了羧酸气相酮基化反应过程, 综述了国内外的研究进展, 并探讨了未来的发展方向. 2013年, Resasco等[38]从反应机理、催化剂和酮基化在生物质转化中的应用三方面进行了系统的报道. Pacchioni等[39]从密度泛函理论(DFT)方面介绍了氧化钛和氧化锆催化羧酸酮基化反应.
不同于之前的综述报道, 本文重点从金属氧化物催化羧酸酮基化反应, 介绍氧化物催化反应机理和催化剂结构性质对酮基化反应的影响这两方面进行论述.
2 金属氧化物催化的酮基化反应机理
尽管酮基化反应经历了很长时间的研究, 但到目前对于酮基化反应机理的认识还没有达成统一.酮基化反应分为体相酮基化反应和表面酮基化反应, 也可以通过生成的中间产物不同将反应机理分为酮烯机理和β-酮酸机理.
2.1 体相与表面酮基化反应
体相酮基化反应是羧酸盐经过裂解生成酮, 具有低晶格能量的氧化物会发生体相反应(如碱金属和碱土金属); 表面酮基化反应则是吸附在催化剂表面的羧化物在α-H的作用下形成不同的中间产物, 进而脱羧生成酮, 具有高晶格能量的氧化物发生表面酮基化反应(如氧化钛、氧化铈、氧化锆、氧化锰)[38].虽然能根据金属氧化物催化剂具有的晶格能量将反应区分为体相进行和表面进行, 但是否经过体相或是表面反应, 不仅与金属氧化物具有的晶格能量有关, 还与反应温度有关.
Zaki等[40]对氧化镁(MgO)研究发现, 虽然MgO具有高晶格能量, 也能进行体相酮基化反应, 这取决于反应温度高低.由于MgO具有强碱性, 在低温条件下, 乙酸分子吸附到催化剂表面形成体相的乙酸镁, 温度高于225 ℃后, 乙酸镁发生热解反应生成丙酮、CO2和H2O.当温度高于300 ℃后, 乙酸在MgO表面活化经过表面酮基化反应生成丙酮、CO2和H2O. Shanks等[31]对氧化铈(CeO2)催化羧酸酮基化反应也做了相同的研究.不管是液相反应还是气相反应, CeO2催化羧酸反应根据反应温度的高低也可以分别进行表面和体相反应.在150~300 ℃之间, CeO2催化羧酸反应是经过体相转化的, 也就是先生成体相羧酸盐, 然后经过热解生成酮、CO2和H2O. Sato等[27]研究了稀土氧化物(氧化镧、氧化铈、氧化镨和氧化钕)催化乙酸酮基化反应, 反应过程中也经过体相转化.体相酮基化反应是通过形成体相羧酸盐发生热解生成酮, 而表面酮基化反应则是在催化剂表面进行.通过在催化剂表面生成的中间物不同又可将酮基化反应机理划分为不同的反应机理, 在2.2和2.3节将详细介绍两种表面催化酮基化反应机理.
2.2 β-酮酸机理
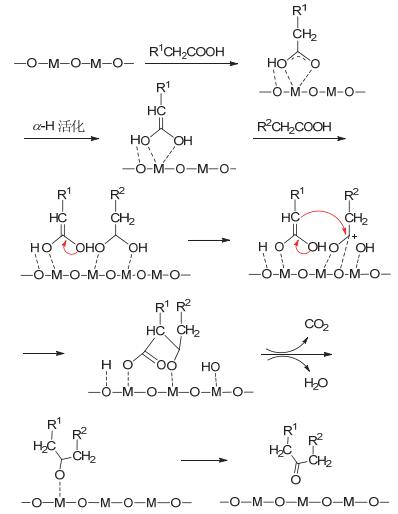
β-酮酸机理是在反应过程中, 两分子羧酸先生成β-酮酸中间产物, 然后再继续转化形成酮, 如图 1所示, 以双配位吸附方式为例.
图 1
金属氧化物催化羧酸酮基化反应机理(β-酮酸机理)示意图
Figure 1.
β-ketoacid based mechanism for ketonization of carboxylic acid over metal oxide
在此反应过程中, 第一个羧酸分子吸附到催化剂表面, 经去质子化后, 羧基的氧原子吸附在金属阳离子上, 而氢原子吸附在表面的氧原子上, 然后α-H经表面的氧原子活化形成羧酸的阴离子或经过烯醇化形成中间物.第二个羧酸分子吸附时, 羧基的氧原子吸附在金属阳离子上, 然后羧基中的碳-羟基键(C—OH键)断裂, 形成乙酰基碳正阳离子.第一个羧酸变形后的中间物亲核进攻酰阳离子, 形成β-酮酸, 然后碳-碳键(C—C键)断裂, 脱去一分子CO2, 形成丙酮; 同时羟基和氢原子键合, 脱去一分子水.金属阳离子吸附羧化物的形式有多种, 单配位吸附、桥连吸附和螯合配位吸附.
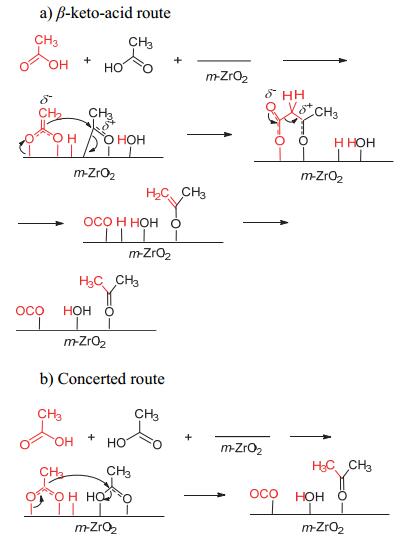
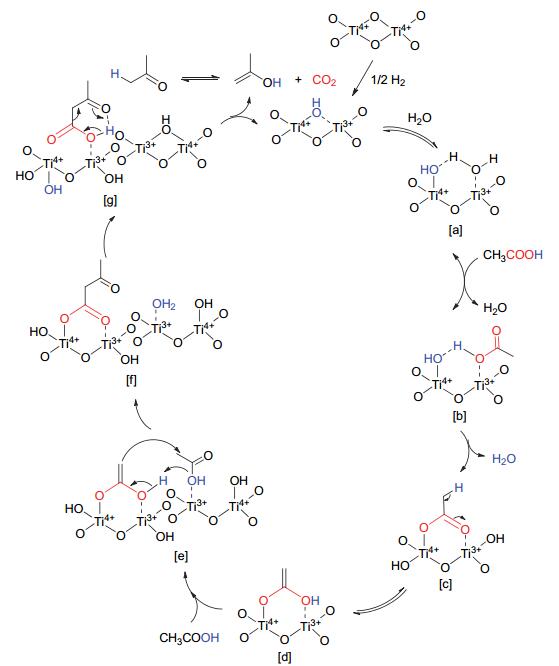
Pulido等[29]通过实验数据并结合DFT计算对比氧化锆(ZrO2)催化乙酸酮基化反应, 此反应可经过β-酮酸过程和直接转化过程, 如图 2所示.在β-酮酸过程中, 两分子乙酸吸附在相邻的活性位上, 一分子乙酸形成烯醇化合物, 另一分子乙酸则形成酰阳离子, 然后烯醇化合物进攻酰阳离子生成β-酮酸中间产物, 然后经过C—C键断裂形成丙酮并脱除CO2和H2O.而在直接转化的路径中, 一分子乙酸盐直接进攻另一分子乙酸, 生成丙酮.经过β-酮酸机理的活化能(108 kJ/mol)要低于直接转化的活化能(154 kJ/mol), 在动力学上更支持β-酮酸机理. Resasco等[41]报道了Ru/TiO2/C催化乙酸发生液相酮基化反应中经过β-酮酸机理, 如图 3所示.在反应之前, 氧化钛先经过H2预处理, 使Ti4+被还原成Ti3+的同时, 表面形成羟基(-OH), 然后进行酮基化反应.在反应中, 一分子乙酸羧基中H与表面羟基键合脱水, 同时乙酸盐中的两个氧原子以桥联的方式吸附在阳离子上, 然后在α-H作用下形成烯醇化合物.之后第二分子乙酸羟基中的氧原子进攻烯醇化合物中的H, 形成β-酮酸中间产物, 再经过C—C键断裂形成丙酮和CO2.虽然二者都提出了β-酮酸机理, 但因为所用催化剂的酸碱性、氧化还原性和表面结构不同导致具体的反应细节不同.在氧化锆的表面, 乙酸可以经过脱羟基形成酰阳离子, 而在氧化钛表面则没有形成.目前大多数的研究者认为羧酸酮基化反应遵循β-酮酸机理[9, 15, 19, 29~31].
2.3 酮烯机理
除了β-酮酸机理外, 也有研究者提出酮烯机理, 但只有具有α-H的羧酸才能经过此过程.酮烯机理就是单分子羧酸先进行脱水生成酮烯(R2C=C=O), 然后再进行C—C耦合反应, 如图 4所示.
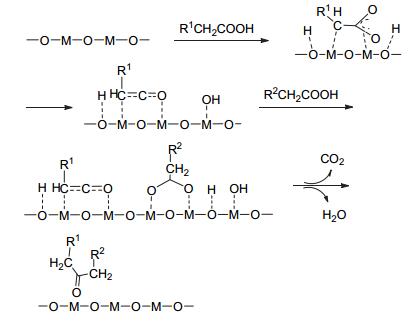 图 4
金属氧化物催化羧酸酮基化反应机理(酮烯机理)示意图
Figure 4.
Ketene-based mechanism of carboxylic acid ketonization over metal oxide
图 4
金属氧化物催化羧酸酮基化反应机理(酮烯机理)示意图
Figure 4.
Ketene-based mechanism of carboxylic acid ketonization over metal oxide
在此反应过程中, 吸附的羧酸分子先进行单分子脱水, 生成酮烯; 然后酮烯直接和羧化物反应生成丙酮和CO2, 或者酮烯转变成乙酰基碳正离子再与羧化物反应生成丙酮和CO2.酰阳离子的形成与氧化物的路易斯酸性有关, 酸性强则有利于形成酰阳离子.
Rajadurai等[35]研究认为羧酸进行酮基化反应是经过酮烯反应机理, 反应式如(2) 所示.当反应温度高于400 ℃时, 吸附的两分子乙酸, 先进行解离生成羧化物(CH3COO-)和氢离子(H+), 然后直接进行酮基化反应; 当反应温度低于400 ℃时, 经过酮烯中间产物生成丙酮. Barteau等[42]使用Pd/CeO2和Co/CeO2低温催化酮基化反应, 中间过程就是乙酰基参与反应生成酮. Dooley等[12]通过对CeO2催化酸-酸、酸-醛的交叉酮基化动力学研究, 采用同位素标记的方法测定中间物, 认为此交叉酮基化反应是经过酮烯机理生成酮.而Ponec等[18]则认为酮烯只是一个副产物而不是中间产物.
3 酮基化金属氧化物催化剂
用于催化酮基化反应的催化剂种类繁多, 有金属氧化物[43, 44]、沸石分子筛[25~27]、Zn/Al氢氧化物[23]、Mg/Al水滑石[24]等.本文聚焦探讨金属氧化物对酮基化反应的影响, 包括催化剂结构、酸碱性、氧化还原性等对反应活性的影响.在过去的几十年里, 开发了大量的金属氧化物用于催化羧酸酮基化反应. 2014年, Glinski等[45]报道了32种元素氧化物对丙酸酮基化反应活性的影响.通过活性测定将氧化物分成三组:低活性催化剂、中等水平活性催化剂、高活性催化剂, 具有高活性的元素包括锰、锆、铈、钍和铀.除了这些负载型的氧化物(载体一般为二氧化硅、三氧化二铝和氧化钛), 混合氧化物(如CeZrOx[46], ZnxZryOz[47], CeMnOx[48]和Zr-Mg-Y-O[49]等)同样具有较高的催化活性.作者采用红外光谱(IR)、程序升温还原(TPR)、程序升温脱附(TPD)、比表面积测定(BET)、X衍射光谱(XRD)等不同的技术表征手段探究催化剂的活性位、结构、酸碱性、氧化还原性对反应活性的影响.本文将从表面结构、酸碱性、氧化还原性这三方面系统介绍金属氧化物催化剂对羧酸酮基化反应的影响.
3.1 催化剂表面结构影响
多相催化反应可以分为两类[50]:一是表面结构敏感性反应, 催化剂的活性取决于催化剂的表面结构, 如晶粒的某一晶面; 二是结构非敏感性反应, 催化剂的活性位与表面结构无关, 此类反应活性可能与催化剂的比表面积有关.对于羧酸酮基化反应, 研究者们发现催化剂的表面结构对反应活性有一定的影响, 如暴露晶面中阴阳离子的配位数.
因其高效的储氧能力和稳定的表面结构, CeO2作为催化材料被广泛应用于多种有机反应中, 如CO氧化[51], 水煤气变换, C—C耦合反应等.在羧酸酮基化反应中, 也被广泛研究. CeO2具有萤石结构, 四价铈阳离子(Ce4+)与八个氧负离子(O2-)配位, 具有多种晶面[52].在CeO2的(111)、(110) 和(100) 晶面中, (111) 晶面最稳定, 也只有(111) 晶面能够催化羧酸酮基化反应[53]. (111) 晶面是六方对称结构, 最外层是O2-离子, 第二层是Ce4+阳离子, 同时暴露在表面, 每一个阳离子和阴离子都有一个空位. (100) 晶面是四方对称结构, 阴阳离子同时暴露在表面, O2-离子是完全配位, Ce4+阳离子则具有四个空位.当阴阳离子同时暴露在表面时, 且金属阳离子具有不饱和配位, 才能吸附解离后的羧化物, 进而经过一系列的转化脱羧生成酮.通过调变催化剂合成方法和焙烧温度等促使形成不同的晶面和不同的颗粒尺寸, 从而控制羧酸酮基化反应的活性.
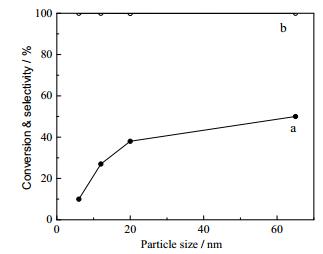
早在1996年, Vohs等[53]报道了CeO2不同晶面对羧酸酮基化反应的影响.研究结果显示, 只有(111) 晶面上能生成酮.结合红外数据, 证明乙酸和甲酸以单配位的形式吸附在Ce4+, 而(100) 晶面存在较多的空位不利于发生酮基化反应.通过改变催化剂合成条件, 可以控制催化剂的颗粒尺寸. Wang等[54]研究了通过控制水热合成条件调变CeO2的颗粒尺寸, 当颗粒大小为3~10 nm时, 主要是(111) 晶面和(100) 晶面.随着颗粒的增大, (100) 晶面逐渐消失, 而(111) 晶面增多, 催化活性增强.焙烧温度同样会影响CeO2颗粒的形貌、结晶性和氧化态, 间接地影响了羧酸酮基化反应活性.不同的焙烧温度会促使形成不同的晶面. Shanks等[55]做了焙烧温度对催化活性的研究, 在450 ℃焙烧时形成的(100) 晶面较多, 而900 ℃焙烧时形成的(111) 晶面则占绝大部分.在液相反应中CeO2催化乙酸酮基化, CeO2的结晶性影响催化活性.随着焙烧温度的升高, 乙酸酮基化初始反应速率逐渐增大, 如表 1所示.在270 ℃反应, 反应速率从52 μmol•m-2•h-1 (450 ℃焙烧)增大到267 μmol•m-2• h-1 (900 ℃焙烧). Sato等[50]研究了在350 ℃丙酸发生酮基化反应, CeO2颗粒尺寸能够影响反应活性, 如图 3所示.随着颗粒尺寸的增大, 丙酸的转化率呈现出增大的趋势, 最高达49.7%.主要产物3-戊酮的选择性都在99%以上, 3-戊酮的时空收率也增加了5倍.
因羧酸酮基化反应活性与氧化物的表面结构息息相关, 氧化钛(TiO2)作为具有三种晶体结构的氧化物被应用到酮基化反应中具有一定的研究意义. TiO2作为催化剂被使用最多的结构是锐钛矿型和金红石型, 二者因其相异的物理、化学性质而有着不同的用途[56].在这两种晶相中, 四价钛阳离子(Ti4+)与六个O2-阴离子配位形成八面体[52]. TiO2的晶面结构和组成决定着羧酸的不同反应路径, 不同晶面对羧酸的吸附方式不同.在还原态的晶面, 羧酸吸附到氧空位上, 直接进行脱氧反应.当晶面上的Ti4+存在不饱和配位时, 则会吸附羧酸发生单分子脱水反应或双分子酮基化反应.
关于锐钛矿TiO2单晶表面催化羧酸反应的研究, Barteau等[57]进行了最早最全面的报道, 探究羧酸反应活性和活性位的关系.在低温时, 乙酸以分子的形式或是解离的形式被吸附在催化剂表面且发生脱附.在高温时, 乙酸盐经过三条不同的路径进行反应, 这取决于催化剂的结构与组成.在还原态的(001) 晶面, 乙酸盐直接进行脱氧反应. (011) 晶面暴露的Ti4+与氧原子是五配位, 单分子乙酸盐吸附在此面直接进行脱水反应. (114) 晶面, 暴露的Ti4+与氧原子是四配位, 双分子乙酸盐吸附在此面进行酮基化反应生成丙酮.在这三种晶面结构中, 只有在(114) 晶面可以进行酮基化反应, 两个乙酸分子以单配位的形式被吸附在同一个Ti4+上. Ojamäe等[58]对锐钛矿TiO2纳米颗粒(110) 晶面催化羧酸反应进行了研究.作者通过红外、拉曼光谱等表征手段, 结合DFT计算, 证明了去质子化后的羧酸两个氧原子吸附在两个Ti4+上, 以桥连的方式存在.催化剂表面的弛豫现象能够影响吸附态的结构和吸附能. Resasco等[19]报道了Ru/TiO2催化羧酸酮基化反应动力学及机理, 也论证了羧酸在催化剂表面以双配位的形式存在.
3.2 催化剂酸碱性影响
金属氧化物表面暴露的晶面类型决定了表面阴阳离子的性质.在金属氧化物中, 暴露的金属阳离子是路易斯酸位(L酸位), O2-离子则是碱性位.金属氧化物催化羧酸酮基化反应的活性位, 一些研究者认为是碱性位[36, 59], 一些人认为酸性位[22]也起着重要的作用.在金属氧化物催化羧酸酮基化反应中, 羧酸先被吸附在催化剂表面进行解离, 羧基的H吸附在O2-, 同时解离后的羧化物吸附在配位不饱和金属阳离子位, 然后α-H经O2-活化, 羧化物经过一系列的变形后与另外一个羧化物进行反应生成酮.在整个反应过程中, 阴阳离子均起到了重要作用, 羧酸酮基化反应需要酸-碱活性位协同作用.
Renz[36]论述了羧酸酮基化反应发生在碱性位. Zaki等[60]利用傅里叶变换红外光谱研究乙酸在Al2O3、CeO2、TiO2这三种氧化物催化下生成丙酮的反应.因Al2O3没有具有可还原的L酸性位, 对丙酸没有催化活性; TiO2和CeO2分别具有可还原的L酸性位(分别是Ti4+和Ce4+), 可分别在400 ℃和300 ℃催化乙酸生成丙酮、CO2和水. CeO2的活性高于TiO2, 是因为CeO2具有更强的碱性. MgO是一种典型的碱性氧化物, 表面暴露弱酸性位(Mg2+)和碱性位(-OH, O2-), 不仅能催化乙酸进行表面的酮基化反应, 还能使乙酸形成体相的乙酸盐, 然后经过裂解生成丙酮, CO2和水[40].金属氧化物催化羧酸酮基化反应, 既需要碱性位活化氢, 同时还需要酸性位吸附羧化物, 这二者缺一不可.
CO2是一种酸性气体, 在反应中占据碱性位, 则通入CO2后反应活性会受到抑制. H2O在催化剂表面分解成氢和羟基, 分别占据了催化剂表面的碱性位和酸性位, 对酮基化反应活性也起到抑制作用. Dumesic等[33]以己酸为模型分子, 研究了CO2和H2O对反应的影响, 研究结果证明了CO2和H2O都起抑制作用.通过NH3-TPD和CO2-TPD测定, ZrO2同时具有酸碱性, 且酸性位数量(212 μmol•gcat-1)与碱性位数量相当(296 μmol•gcat-1).当以原子比1/1掺杂Ce后(Ce0.5Zr0.5O2), 催化剂的碱性位增多(380 μmol•gcat-1), 酸性位减少(56 μmol•gcat-1).此时Ce0.5Zr0.5O2催化活性要高于ZrO2, 碱性位增多有利于提高酮基化反应活性.
Kijenski等[59]研究了二十种负载型氧化物催化乙酸酮基化反应, 如表 2所示.在这些氧化物中, 有碱性氧化物、酸性氧化物和同时具有酸碱性的两性氧化物.研究结果表明, 两性氧化物(CeO2、MnO2、La2O3)的催化活性要高于酸性氧化物和碱性氧化物.
 表 2
金属氧化物对乙酸酮基化反应活性影响[59]
Table 2.
Activity of 10 wt% MOx/SiO2 catalysts in ketonization of acetic acid[59]
表 2
金属氧化物对乙酸酮基化反应活性影响[59]
Table 2.
Activity of 10 wt% MOx/SiO2 catalysts in ketonization of acetic acid[59]
Oxide Yield of acetone/% 300 ℃ 325 ℃ 350 ℃ 375 ℃ 400 ℃ 425 ℃ SiO2 2 2 3 4 5 20 B2O3 2 2 2 2 3 6 MoO3 2 2 1 4 5 5 WO3 2 4 6 6 5 5 P2O5 1 1 6 10 12 9 V2O5 3 4 4 9 21 29 Bi2O3 10 6 5 11 18 28 NiO 7 9 10 31 — — Al2O3 0 0 4 15 37 45 CuO 5 5 6 29 39 43 ZnO 6 9 10 19 33 54 PbO 6 10 15 36 76 79 Cr2O3 1 8 52 48 39 46 Fe2O3 13 32 39 66 59 60 CoO 13 15 48 50 63 64 MgO 7 20 39 53 59 68 Nd2O3 3 3 6 22 61 70 La2O3 3 12 14 50 87 — MnO2 18 22 34 72 96 — CdO 6 27 73 76 94 — CeO2 9 24 31 96 97 — 3.3 催化剂氧化还原性影响
除了对酸碱性的探究, 研究者们也研究了催化剂氧化还原性对羧酸酮基化活性的影响, 尤其是CeO2、TiO2、ZrO2这些易被还原的氧化物.经研究发现, 催化剂经过还原后表面形成氧空位, 暴露了更多的配位不饱和金属阳离子位, 去质子化后的羧化物更容易吸附在配位不饱和金属阳离子位, 有利于反应的进行.
Zaki等[60]研究了Al2O3, CeO2, TiO2三种氧化物催化乙酸酮基化反应.这三种氧化物都具有L酸位, 但只有Al2O3对乙酸酮基化反应没有催化活性, 究其原因是Al2O3的阳离子不能被还原, 不能形成具有配位不饱和金属阳离子位. Resasco等[19, 30, 41]研究了Ru/TiO2催化羧酸酮基化反应.不管是在液相反应还是气相反应中, 催化剂经过还原后对反应的活性均有显著提高.催化剂经过H2还原后, Ti4+被还原成Ti3+, 形成了更多的氧空位、暴露了更多的配位不饱和金属阳离子, 大大地提高了羧酸的转化率. Simakova等[46]在2013年报道了反应气氛对CeO2、ZrO2、CeO2/ZrO2催化戊酸酮基化反应活性的影响, 比较了N2和H2对反应活性的影响(如表 3所示), 通入氢气后催化剂被还原并暴露了更多的金属阳离子, 戊酸的转化率得到显著提高. 2014年, Simakova等[61]又报道了在H2气氛下, ZrO2催化戊酸酮基化反应机理的研究.通过采用红外光谱和紫外-可见漫反射光谱(UV-vis DRS)的表征手段, 探究了ZrO2对戊酸和5-己酮的吸附方式.结合实验数据和DFT计算, 证明ZrO2吸附戊酸盐的方式取决于反应条件.在H2反应气氛下, 四价锆阳离子(Zr4+)可被还原成三价锆阳离子(Zr3+), 戊酸在ZrO2表面的存在方式可能是单分子单配位、单分子桥连或是螯合配位, 也可能是两个分子同时被吸附在一个锆离子上. Overbury等[21]研究了CeO2 (111) 晶面初始氧化状态对乙酸反应的影响.乙酸在CeO2 (111) 晶面反应生成物的脱附情况取决于催化剂表面的初始氧化状态.随着还原程度的增高, 形成的氧空位增多, 乙酸越有利于进行单分子脱氧形成乙醛而不利于进行酮基化形成丙酮.适度还原催化剂有利于提高酮基化活性, 但过度还原则不利于酮基化反应. Pacchioni等[62]则通过密度泛函理论计算研究ZrO2预还原对乙酸酮基化反应的影响.在未还原的表面上, 去质子化后的乙酸盐进行烯醇化过程需要较大的活化能, 并且很难进行脱氧形成乙酰阳离子.而ZrO2经过H2还原处理后, Zr3+周围的电子很容易转移到乙酸盐离子上, 进行脱氧形成乙酰基, 再与烯醇化合物反应生成β-酮酸.同时氧空位的存在也有利于乙酰基中间物的形成.
表 3
H2和N2对CeO2、ZrO2、10% CeO2/ZrO2催化戊酸酮基化活性的影响[46]
Table 3.
Catalytic properties of CeO2, ZrO2 and 10% CeO2/ZrO2 oxides in the ketonization of valeric acid in hydrogen and nitrogen[46]
Sample Conversion/% Selectivity/% S/(m2•g-1) H2 N2 H2 N2 CeO2 40 31 76 80 25 ZrO2 83 66 78 80 105 10%-CeO2/ZrO2 93 82 79 81 79 Reaction conditions: 355 ℃, 1 bar, H2 flow rate 30 cm3•min-1, valeric acid LHSV=0.34 cm3•g-1•h-1. 4 结论与展望
伴随着石化能源的消耗和环境污染的日益严重, 生物质能源作为一种可再生的清洁资源, 越来越引起研究者们的注意.通过一系列的反应过程可将木质纤维素转化成油品和化学品, 来满足人们的需求.羧酸在转化过程中具有较高的含量, 所以将羧酸进行转化是非常关键的一步.研究发现, 通过酮基化反应将有机羧酸转化是一条可行的方法, 但开发出具有低成本、高催化活性的催化剂是在工业应用中要解决的重要的问题, 这也是我们所面临的挑战与机遇.金属氧化物催化羧酸酮基化反应的机理与氧化物的晶格能量和反应温度等有关, 高晶格能量的氧化物在高温下进行表面催化反应, 在低温下进行体相反应.当羧酸进行表面酮基化反应时, 根据羧酸分子的吸附情况不同生成的中间物不同, 可以分为β-酮酸机理和酮烯机理.金属氧化物催化羧酸酮基化反应中, 氧化物表面结构决定羧酸的吸附方式和吸附能, 从而影响反应活性.氧化物酸碱性和还原性的改变直接影响了表面结构的状态, 因此氧化物的酸碱性和还原性也影响着酮基化反应的进行.
酮基化反应既可以在高温下发生气相反应, 也可以在低温下发生液相反应.目前大多数研究者们关注于气相酮基化反应, 对液相反应研究的较少.生物质的衍生物种类繁多、且难于分离, 易溶于水, 所以对酮基化液相反应的研究显得更为有意义.在酮基化反应的研究中, 除了反应机理、催化剂种类及活性位的探索, 催化剂的稳定性和失活的问题也是值得关注的.不管在气相反应还是在液相反应, 催化剂很容易失活.造成失活的原因很多, 如积碳、反应中间物堵塞活性位、溶剂的影响等, 这都需要研究者们进行深入细致的研究.
羧酸酮基化反应属于表面结构敏感型反应, 调控催化剂的表面结构和性质可能会提高催化剂在温和条件下的活性, 是未来值得研究的重要方向之一.选用两性金属氧化物作为主活性组分, 在此基础上掺杂或负载其他金属元素, 来调控催化剂的表面结构(如酸碱对数量和暴露金属阳离子的不饱和配位度)和性质(酸碱性和氧化还原性), 有可能提高催化剂的反应活性, 进而使酮基化反应在更温和条件下进行.
虽然酮基化反应是一个传统的化学反应, 但到目前为止, 众多影响因素对酮基化反应活性的影响没有得到统一的认知.所以基于生物质能源的开发和利用, 对羧酸酮基化反应深入细致的研究还是很有必要的, 且具有重要的意义.
-
-
[1]
Huber, G. W.; Iborra, S.; Corma, A. Chem. Rev. 2006, 106, 4044. doi: 10.1021/cr068360d
-
[2]
Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411. doi: 10.1021/cr050989d
-
[3]
Goyal, H. B.; Seal, D.; Saxena, R. C. Renew. Sust. Energ. Rev. 2008, 12, 504. doi: 10.1016/j.rser.2006.07.014
-
[4]
陶骏骏, 陈帅, 姚奉奇, 王海晖, 化学学报, 2016, 74, 81. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345276.shtmlTao, J. J.; Chen, S.; Yao, F. Q.; Wang, H. H. Acta Chim. Sinica. 2016, 74, 81 (in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345276.shtml
-
[5]
Ou, J. K.; Yang, L.; Xi, X. H. Chin. J. Chem. 2016, 34, 727. doi: 10.1002/cjoc.v34.7
-
[6]
Dai, N.; Shang, R.; Fu, M. C.; Fu, Y. Chin. J. Chem. 2015, 33, 405. doi: 10.1002/cjoc.v33.4
-
[7]
李江, 黄耀兵, 郭庆祥, 傅尧, 化学学报, 2014, 72, 1223. doi: 10.3866/PKU.WHXB201405091Li, J.; Huang, Y. B.; Guo, Q. X.; Fu, Y. Acta Chim. Sinica. 2014, 72, 1223 (in Chinese). doi: 10.3866/PKU.WHXB201405091
-
[8]
Bertero, M.; Puente, G. D. L.; Sedran, U. Fuel. 2012, 95, 263. doi: 10.1016/j.fuel.2011.08.041
-
[9]
Ignatchenko, A. V.; Kozliak, E. I. ACS Catal. 2012, 2, 1555. doi: 10.1021/cs3002989
-
[10]
Nagashima, O.; Sato, S.; Takahashi, R.; Sodesawa, T. J. Mol. Catal. A: Chem. 2005, 227, 231. doi: 10.1016/j.molcata.2004.10.042
-
[11]
Randery, S. D.; Warren, J. S.; Dooley, K. M. Appl. Catal., A 2002, 226, 265. doi: 10.1016/S0926-860X(01)00912-7
-
[12]
Hendren, T. S.; Dooley, K. M. Catal. Today 2003, 85, 333. doi: 10.1016/S0920-5861(03)00399-7
-
[13]
Dooley, K. M.; Bhat, A. K.; Plaisance, C. P.; Roy, A. D. Appl. Catal., A 2007, 320, 122. doi: 10.1016/j.apcata.2007.01.021
-
[14]
Murkute, A. D.; Jackson, J. E.; Miller, D. J. J. Catal. 2011, 278, 189. doi: 10.1016/j.jcat.2010.12.001
-
[15]
Ignatchenko, A. V.; Deraddo, J. S.; Marino, V. J.; Mercado, A. Appl. Catal., A 2015, 498, 10. doi: 10.1016/j.apcata.2015.03.017
-
[16]
Friedel, C. Justus Liebigs Ann. Chem. 1858, 108, 122. doi: 10.1002/(ISSN)1099-0690
-
[17]
胡淼, 朱志庆, 徐泽辉, 化工进展, 2010, 29, 316. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmHu, M.; Zhu, Z. Q.; Xu, Z. H. Chem. Ind. Eng. Prog. 2010, 29, 316 (in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[18]
Pestman, R.; Koster, R. M.; Duijne, A. V.; Pieterse, J. A. Z.; Ponec, V. J. Catal. 1997, 168, 265. doi: 10.1006/jcat.1997.1624
-
[19]
Pham, T. N.; Shi, D.; Resasco, D. E. J. Catal. 2014. 314, 149. doi: 10.1016/j.jcat.2014.04.008
-
[20]
Pei, Z. F.; Ponec, V. Appl. Surf. Sci. 1996, 103, 171. doi: 10.1016/0169-4332(96)00453-9
-
[21]
Calaza, F. C.; Chen, T. L.; Mullins, D. R.; Xu, Y.; Overbury, S. H. Catal. Today 2015, 253, 65. doi: 10.1016/j.cattod.2015.03.033
-
[22]
Lee, Y.; Choi, J. W.; Suh, D. J.; Ha, J. M.; Lee, C. H. Appl. Catal., A 2015, 506, 288. doi: 10.1016/j.apcata.2015.09.008
-
[23]
Das, J.; Parida, K. React. Kinet. Catal. Lett. 2000, 69, 223. doi: 10.1023/A:1005627228083
-
[24]
Parida, K.; Das, J. J. Mol. Catal. A: Chem. 2000, 151, 185. doi: 10.1016/S1381-1169(99)00240-X
-
[25]
Martens, J. A.; Wydoodt, M.; Espeel, P.; Jacobs, P. A. Stud. Surf. Sci. Catal. 1993, 78, 527. doi: 10.1016/S0167-2991(08)63362-5
-
[26]
Gumidyala, A.; Sooknoi, T.; Crossley, S. J. Catal. 2016, 340, 76. doi: 10.1016/j.jcat.2016.04.017
-
[27]
Yamada, Y.; Segawa, M.; Sato, F.; Kojima, T.; Sato, S. J. Mol. Catal. A: Chem. 2011, 346, 79. doi: 10.1016/j.molcata.2011.06.011
-
[28]
孙春晖, 陈永生, 李佳, 无机盐工业, 2008, 40, 44. doi: 10.3969/j.issn.1006-4990.2008.03.015Sun, C. H.; Chen, Y. S.; Li, J. Inorg. Chem. Ind. 2008, 40, 44 (in Chinese). doi: 10.3969/j.issn.1006-4990.2008.03.015
-
[29]
Pulido, A.; Oliver-Tomas, B.; Renz, M.; Boronat, M.; Corma, A. Chem Sus Chem 2013, 6, 141. doi: 10.1002/cssc.201200419
-
[30]
Pham, T. N.; Dachuan, S.; Resasco, D. E. Top. Catal. 2014, 57, 706. doi: 10.1007/s11244-013-0227-7
-
[31]
Snell, R. W.; Shanks, B. H. ACS Catal. 2013, 3, 783. doi: 10.1021/cs400003n
-
[32]
Gaertner, C. A.; Serrano-Ruiz, J. C.; Braden, D. J.; Dumesic, J. A. Ind. Eng. Chem. Res. 2010, 49, 6023.
-
[33]
Gaertner, C. A.; Serrano-Ruiz, J. C.; Braden, D. J.; Dumesic, J. A. J. Catal. 2009, 266, 71. doi: 10.1016/j.jcat.2009.05.015
-
[34]
Gaertner, C. A.; Serrano-Ruiz, J. C.; Braden, D. J.; Dumesic, J. A. Chem Sus Chem 2009, 2, 1121. doi: 10.1002/cssc.v2:12
-
[35]
Rajadurai, S. Catal. Rev. 1994, 36, 385. doi: 10.1080/01614949408009466
-
[36]
Renz, M. Eur. J. Org. Chem. 2005, 2005, 979. doi: 10.1002/(ISSN)1099-0690
-
[37]
宁朋歌, 曹宏斌, 张懿, 现代化工, 2008, 28, 22. doi: 10.3321/j.issn:0253-4320.2008.01.006Ning, P. G.; Cao, H. B.; Zhang, Y. Mod. Chem. Ind. 2008, 28, 22 (in Chinese). doi: 10.3321/j.issn:0253-4320.2008.01.006
-
[38]
Pham, T. N.; Tawan, S.; Steven, P. C.; Resasco, D. E. ACS Catal. 2013, 3, 2456. doi: 10.1021/cs400501h
-
[39]
Pacchioni, G. ACS Catal. 2014, 4, 2874. doi: 10.1021/cs500791w
-
[40]
Mekhemer, G. A. H.; Halawy, S. A.; Mohamed, M. A.; Zaki, M. I. J. Catal. 2005, 230, 109. doi: 10.1016/j.jcat.2004.09.030
-
[41]
Pham, T. N.; Shi, D.; Sooknoi, T.; Resasco, D. E. J. Catal. 2012, 295, 169. doi: 10.1016/j.jcat.2012.08.012
-
[42]
Idriss, H.; Diagne, C.; Hindermann, J. P.; Kiennemann, A.; Barteau, M. A. J. Catal. 1995, 155, 219. doi: 10.1006/jcat.1995.1205
-
[43]
张义, 高中良, 陈永生, 李煦, 稀有金属, 2010, 34, 574. doi: 10.3969/j.issn.0258-7076.2010.04.019Zhang, Y.; Gao, Z. L.; Chen, Y. S.; Li, X. Chin. J. Rare Met. 2010, 34, 574 (in Chinese). doi: 10.3969/j.issn.0258-7076.2010.04.019
-
[44]
张义, 高中良, 陈永生, 李煦, 无机盐工业, 2010, 4, 33. doi: 10.3969/j.issn.1006-4990.2010.04.011Zhang, Y.; Gao, Z. L.; Chen, Y. S.; Li, X. Inorg. Chem. Ind. 2010, 4, 33 (in Chinese). doi: 10.3969/j.issn.1006-4990.2010.04.011
-
[45]
Gliński, M.; Zalewski, G.; Burno, E.; Jerzak, A. Appl. Catal., A 2014, 470, 278. doi: 10.1016/j.apcata.2013.10.047
-
[46]
Zaytseva, Y. A.; Panchenko, V. N.; Simonov, M. N.; Shutilov, A. A.; Zenkovets, G. A.; Renz, M.; Simakova, I. L.; Parmon, V. N. Top. Catal. 2013, 56, 846. doi: 10.1007/s11244-013-0045-y
-
[47]
Crisci, A. J.; Dou, H.; Prasomsri, T.; Román-Leshkov, Y. ACS Catal. 2014, 4, 4196. doi: 10.1021/cs501018k
-
[48]
Snell, R. W.; Shanks, B. H. ACS Catal. 2014, 4, 512. doi: 10.1021/cs400851j
-
[49]
Teterycz, H.; Klimkiewicz, R.; Łaniecki, M. Appl. Catal., A 2003, 249, 313. doi: 10.1016/S0926-860X(03)00231-X
-
[50]
Kobume, M.; Sato, S.; Takahashi, R. J. Mol. Catal. A: Chem. 2008, 279, 10. doi: 10.1016/j.molcata.2007.09.027
-
[51]
Wang, W. D.; Lin, P. Y.; Fu, Y. L.; Yu, S. M.; Meng, M.; Zhang, X. P. Chin. J. Chem. 2000, 18, 673.
-
[52]
Vohs, J. M. Chem. Rev. 2013, 113, 4136. doi: 10.1021/cr300328u
-
[53]
Stubenrauch, J.; Brosha, E.; Vohs, J. M. Catal. Today 1996, 28, 431. doi: 10.1016/S0920-5861(96)00251-9
-
[54]
Wang, Z. L.; Feng, X. D. J. Phys. Chem. B 2003, 107, 13563. doi: 10.1021/jp036815m
-
[55]
Snell, R. W.; Shanks, B. H. Appl. Catal., A 2013, 451, 86. doi: 10.1016/j.apcata.2012.08.043
-
[56]
孙静, 高濂, 化学学报, 2002, 60, 1524. doi: 10.3321/j.issn:0567-7351.2002.08.030Sun, J.; Gao, L. Acta Chim. Sinica 2002, 60, 1524 (in Chinese). doi: 10.3321/j.issn:0567-7351.2002.08.030
-
[57]
Kim, K. S.; Barteau, M. A. J. Catal. 1990, 125, 353. doi: 10.1016/0021-9517(90)90309-8
-
[58]
Ojamäe, L.; Aulin, C.; Pedersen, H.; Käll, P. O. J. Colloid Interface Sci. 2006, 296, 71. doi: 10.1016/j.jcis.2005.08.037
-
[59]
Glinski, M.; Kijenski, J.; Jakubowski, A. Appl. Catal., A 1995, 128, 209. doi: 10.1016/0926-860X(95)00082-8
-
[60]
Hasan, M. A.; Zaki, M. I.; Pasupulety, L. Appl. Catal., A 2003, 243, 81. doi: 10.1016/S0926-860X(02)00539-2
-
[61]
Panchenko, V. N.; Zaytseva, Y. A.; Simonov, M. N.; Simakova, I. L.; Paukshtis, E. A. J. Mol. Catal. A: Chem. 2014, 388, 133.
-
[62]
Tosoni, S.; Pacchioni, G. J. Catal. 2016, 344, 465. doi: 10.1016/j.jcat.2016.10.002
-
[1]
-

图 1 金属氧化物催化羧酸酮基化反应机理(β-酮酸机理)示意图
Figure 1 β-ketoacid based mechanism for ketonization of carboxylic acid over metal oxide



图 4 金属氧化物催化羧酸酮基化反应机理(酮烯机理)示意图
Figure 4 Ketene-based mechanism of carboxylic acid ketonization over metal oxide

Catalyst Tcalcination/℃ Initial rate/(μmol•m-2•h-1), 270 ℃ Initial rate/(μmol•m-2•h-1), 290 ℃ 450 52 333 600 94 408 750 258 835 900 267 1484  下载: 导出CSV
下载: 导出CSV
表 2 金属氧化物对乙酸酮基化反应活性影响[59]
Table 2. Activity of 10 wt% MOx/SiO2 catalysts in ketonization of acetic acid[59]
Oxide Yield of acetone/% 300 ℃ 325 ℃ 350 ℃ 375 ℃ 400 ℃ 425 ℃ SiO2 2 2 3 4 5 20 B2O3 2 2 2 2 3 6 MoO3 2 2 1 4 5 5 WO3 2 4 6 6 5 5 P2O5 1 1 6 10 12 9 V2O5 3 4 4 9 21 29 Bi2O3 10 6 5 11 18 28 NiO 7 9 10 31 — — Al2O3 0 0 4 15 37 45 CuO 5 5 6 29 39 43 ZnO 6 9 10 19 33 54 PbO 6 10 15 36 76 79 Cr2O3 1 8 52 48 39 46 Fe2O3 13 32 39 66 59 60 CoO 13 15 48 50 63 64 MgO 7 20 39 53 59 68 Nd2O3 3 3 6 22 61 70 La2O3 3 12 14 50 87 — MnO2 18 22 34 72 96 — CdO 6 27 73 76 94 — CeO2 9 24 31 96 97 —
下载: 导出CSV
表 3 H2和N2对CeO2、ZrO2、10% CeO2/ZrO2催化戊酸酮基化活性的影响[46]
Table 3. Catalytic properties of CeO2, ZrO2 and 10% CeO2/ZrO2 oxides in the ketonization of valeric acid in hydrogen and nitrogen[46]
Sample Conversion/% Selectivity/% S/(m2•g-1) H2 N2 H2 N2 CeO2 40 31 76 80 25 ZrO2 83 66 78 80 105 10%-CeO2/ZrO2 93 82 79 81 79 Reaction conditions: 355 ℃, 1 bar, H2 flow rate 30 cm3•min-1, valeric acid LHSV=0.34 cm3•g-1•h-1.
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 38
- 文章访问数: 2992
- HTML全文浏览量: 595
 下载:
下载:
