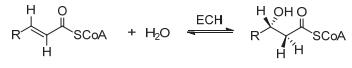
 图 1
ECH催化的α, β-不饱和硫酯水合反应
Figure 1.
Hydration of α, β-unsaturated thiolester catalyzed by ECH
图 1
ECH催化的α, β-不饱和硫酯水合反应
Figure 1.
Hydration of α, β-unsaturated thiolester catalyzed by ECH
引用本文:
章瑜, 杨新亚, 于海瀛, 马广才. 烯酰基-辅酶A水合酶催化机理的理论研究[J]. 化学学报,
2017, 75(5): 494-500.
doi:
10.6023/A16100559

Citation: Zhang Yu, Yang Xinya, Yu Haiying, Ma Guangcai. Theoretical Insight into the Catalytic Mechanism of Enoyl-CoA Hydratase[J]. Acta Chimica Sinica, 2017, 75(5): 494-500. doi: 10.6023/A16100559
Citation: Zhang Yu, Yang Xinya, Yu Haiying, Ma Guangcai. Theoretical Insight into the Catalytic Mechanism of Enoyl-CoA Hydratase[J]. Acta Chimica Sinica, 2017, 75(5): 494-500. doi: 10.6023/A16100559
烯酰基-辅酶A水合酶催化机理的理论研究
摘要:
采用DFT方法研究了烯酰基-辅酶A(ECH)催化的4-(N,N-二甲氨基)-肉桂酰-辅酶A(DAC-CoA)和巴豆酰基-辅酶A(Crotonyl-CoA)水合反应.计算表明:水合反应以分步机理进行,经历一个烯醇负离子中间体.Glu164残基作为唯一的催化碱/酸参与水合反应,而Glu144虽然没有直接参与反应,但是它能通过氢键作用诱使水分子以合适的朝向活化底物.Crotonyl-CoA底物的水加成活性高于DAC-CoA.Ala98和Gly141与底物羰基之间的氢键作用既有利于底物的准确结合,也能有效稳定反应中形成的过渡态和中间体.另外,Glu144和Glu164周围的氢键网络对于合理维持活性位点排布进而有效促进底物活化也很重要.
-
关键词:
- 烯酰基-辅酶A水合酶
- / 催化机理
- / 密度泛函
- / 4-(N, N-二甲氨基)-肉桂酰-辅酶A
- / 巴豆酰基-辅酶A
English
Theoretical Insight into the Catalytic Mechanism of Enoyl-CoA Hydratase
Abstract:
Enoyl-CoA hydratase (ECH), which is also known as crotonase, is the second requisite enzyme in the β-oxidation pathway of fatty acid that catalyzes the syn hydration of α, β-unsaturated thiolester substrates. In this work, ECH-catalyzed hydration mechanisms of DAC-CoA and Crotonyl-CoA were investigated using density functional theory (DFT) methods. Geometrical structures were optimized using Gaussian 03 program at the B3LYP/6-31G(d, p) level of theory. Frequency calculations were performed with the 6-31G(d, p) basis set to obtain zero-point vibrational energies (ZPEs) and to confirm the nature of all the stationary points that have no imaginary frequency for the local minima and have only one imaginary frequency for the saddle points. The single-point calculations on the optimized geometries were further performed with 6-311++G(2d, 2p) basis set to obtain more accurate energies. The polarizable-continuum model (PCM) with the dielectric constant of 4 was used to calculate the single point energies at 6-311++G(2d, p) level on all the optimized geometries to consider the effects of enzymatic environment that was not included in the computational model. Considering that B3LYP functional lacks the proper description of the long-range dispersion interactions, we further used the DFT-D3 program to calculate the empirical dispersion correction to correct the B3LYP energies. The final energies reported in this work are the single-point energies corrected for ZPEs, solvation and dispersion effects. The calculated results suggested that hydration proceeds through a stepwise mechanism, involving an enolate intermediate. Glu164 functions as the sole base/acid for catalysis. Although Glu144 is not directly involved in hydration, it induces the catalytic water molecule to locate an ideal orientation to attack the double bond of substrate by the hydrogen-bonding interaction. Crotonyl-CoA shows higher hydration activity than DAC-CoA. The backbone NH groups of Ala98 and Gly141 form an oxyanion hole with substrate carbonyl oxygen, which play key roles in binding substrate and stabilizing the generated transition states and intermediates. In addition, the hydrogen-bonding networks surrounding Glu144 and Glu164 are of great importance for active site arrangement.
-
Key words:
- Enoyl-CoA hydratase
- / catalytic mechanism
- / density functional theory
- / DAC-CoA
- / Crotonyl-CoA
-
1 引言
烯酰基-辅酶A水合酶(enoyl-CoA hydratase, ECH)也被称作巴豆酸酶(crotonase), 是脂肪酸β-氧化代谢路径中所必需的一种酶, 它催化反式α, β-不饱和硫酯的双键基团发生syn水加成反应生成(S)-3-羟基酰基-辅酶A硫酯产物(图 1)[1].不过有研究表明ECH也能催化顺式α, β-不饱和硫酯水合[2].在有机体内, 水合酶超家族催化的双键水合是很常见的反应[3, 4], 而且α, β-不饱和酮的不对称共轭水加成是一种重要的对映选择性的生物转化过程[5, 6].近年来, 不同物种的ECH X-射线晶体结构已经被广泛报道[7~11].结构分析表明ECH通常以六聚体的形式存在, 由两个完全相同三聚体堆积而成, 且每个亚基都包含一个有催化功能的活性位点[8~10].
图 1
ECH催化的α, β-不饱和硫酯水合反应
Figure 1.
Hydration of α, β-unsaturated thiolester catalyzed by ECH
早期的实验研究已经对ECH催化机理及活性位点残基的作用进行了预测.残基突变研究表明, 在老鼠肝组织ECH中, 残基Glu144和Glu164在催化反应中起关键作用(图 2)[8, 12, 13].这两个残基协同作用, 作为催化碱直接活化活性位点中的水分子以启动水合反应.此外, 结构分析表明Ala98和Gly141骨架酰胺基团可以通过氢键相互作用与底物羰基形成一个“氧负离子穴(oxyanion hole)”, 它能够合理定位底物烯酰基部分的朝向, 而且突变试验进一步证实它能有效促进酶催化作用[7, 8, 12, 14].氨基酸序列分析表明在ECH超酶家族中Gly141是一个高度保守的残基, 而把Gly141突变成Pro会显著降低酶活性[12, 15]. Bahnson等[8]首次报道了ECH结合底物4-(N, N-二甲氨基)-肉桂酰-辅酶A (DAC-CoA)的晶体结构(图 2).随后, D'Ordine等[16]通过13C NMR光谱分析给出了清晰的酶-底物复合物构象, 并通过量子化学计算和电子应变能计算进一步分析了肉桂酰-辅酶A底物在ECH活性位点中的电子极化作用.底物的极化电子可以与酶活性位点氨基酸形成一个静电场, 进而有效促进酶催化反应.早期的动力学同位素效应实验建议了一个协同的双键水合机理, 反应中C—OH键和C—H键的形成只需一个化学步骤[3, 17].然而, 随后的实验研究及理论计算建议ECH催化的α, β-不饱和硫酯水合更可能以分步机理进行, 反应会经历一个烯醇负离子中间体[8, 18].此外, 研究还发现水合产物中Cα位的H来自于亲核进攻底物双键的活性水分子, 而不是来自于任何一个活性位点极性氨基酸[8].
尽管ECH催化机理已经被广泛研究, 但是一些关键性的问题依然悬而未决.例如:哪一个活性位点谷氨酸残基才是真正的催化碱, 亦或两个都有可能?水加成反应到底是协同的还是分步的?反应的具体细节及能量关系如何?现有的实验结果并不能圆满地回答这些关键问题, 因此就需要借助理论计算从微观水平上深入探讨ECH催化α, β-不饱和硫酯水合的详细机理.最近, Cui等[19]使用DFT方法考察了Glu144和Glu164不同质子化状态下的DAC-CoA水合活性, 研究发现Glu144和Glu164都应该以去质子化的状态存在.不过, 严格来说由于共轭π键的存在, DAC-CoA并不是ECH催化的理想底物, 不然实验上也很难得到ECH-DAC-CoA复合物的晶体结构.实验研究表明, 活性最高的底物是巴豆酰基-辅酶A (crotonyl-CoA), 而且随着底物链长度的增加, ECH催化的转化数会急剧降低[20].因此, 通过理论计算研究crotonyl-CoA水合机理对于准确探讨ECH的催化活性至关重要.
近年来, DFT计算方法已经被广泛且成功地应用于酶催化反应机理研究[21~25].在本文中, 我们采用DFT方法探讨了ECH催化的DAC-CoA和Crotonyl-CoA水合反应.通过DFT计算, 我们确定了最优反应路径及反应的势能面, 比较了两种底物的水合活性, 明确了一些活性位点氨基酸的重要作用, 并揭示了不同大小的计算模型对反应势能面的影响.
2 计算方法与模型
2.1 计算方法
所有的理论计算采用杂化B3LYP泛函[26, 27], 由Gaussian03程序包[28]完成.构型优化使用6-31G(d, p)基组.为了获得更准确的能量数据, 在优化的构型的基础上使用更高水平的6-311++G(2d, 2p)基组进行单点能计算.同时, 在单点能计算中使用极化连续介质模型(PCM)[29, 30]计算溶剂化效应以模拟真实的酶环境对反应能垒的影响, 其中酶环境的介电常数(ε)设定为4, 该值能比较理想地模拟蛋白质本身(介电常数约为3) 和蛋白质周围的水介质(介电常数约为80) 的平均极化效应.频率计算使用6-31G(d, p)基组完成以得到零点能(ZPE).考虑到B3LYP泛函不能准确地描述长程色散相互作用, 我们进一步使用DFT-D3方法[31, 32]矫正了B3LYP能量.文中报道的所有能量都是进行溶剂化效应、色散效应和ZPE校正后的单点能.在构型优化中, 氨基酸残基中的一些原子将会被固定在它们结晶时的原始位置以防止不合理的移动.这种处理会导致优化的构型出现一些小虚频, 通常小于50i cm-1.这些小虚频对零点能影响很小, 因此我们可以忽略不计.
2.2 活性位点模型
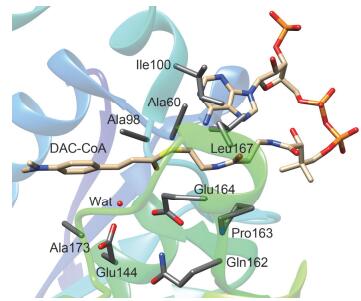
基于ECH结合底物DAC-CoA的活性位点结构(PDB编码: 1EY3, 图 2)[8]构建计算模型.结构分析表明, ECH是由两个完全相同的三聚体堆积而成的六聚体复合物, 其中每个亚基单体包含一个独立的活性位点, 且所有的活性位点都相同.因此, 我们选择其中一个亚基的活性位点构建计算模型. 图 3显示了构建的ECH活性位点模型示意图.我们先用小模型研究DAC-CoA和Crotonyl-CoA水合机理, 包括底物α, β-不饱和硫酯部分、Wat、Glu144、Glu164以及Ala98、Gly141和Gly172的骨架酰胺基团.随后, 为了更好的模拟ECH催化活性, 我们进一步把Gln162及Glu164、Ala173和Gly175的酰胺基团纳入计算模型.氨基酸和底物的截断处如图 3所示, 所有的氢原子都手动添加.在构型优化过程中, 截断处的C原子固定在晶体中的原始位置保持不动以避免基团的不合理运动, 且为了便于观察, 所有固定的原子用星号标记.
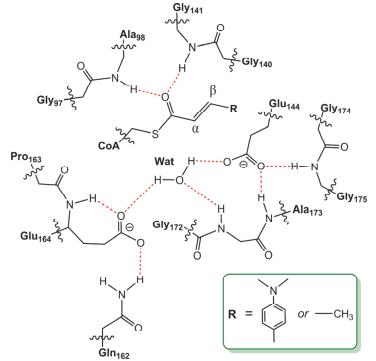 图 3
DFT计算构建的活性位点模型示意图.底物中R可以是4-(N, N-二甲氨基)-苯基或者甲基.红色虚线代表可能的氢键作用
Figure 3.
Constructed active site model for DFT calculations. R group in substrate can be 4-dimethylamino-phenyl or methyl. Red dashed lines represent the possible hydrogen-bonding interactions
图 3
DFT计算构建的活性位点模型示意图.底物中R可以是4-(N, N-二甲氨基)-苯基或者甲基.红色虚线代表可能的氢键作用
Figure 3.
Constructed active site model for DFT calculations. R group in substrate can be 4-dimethylamino-phenyl or methyl. Red dashed lines represent the possible hydrogen-bonding interactions
3 结果与讨论
3.1 DAC-CoA水合反应
我们首先通过DFT计算探讨ECH催化的DAC-CoA水合机理.检查ECH-DAC-CoA复合物晶体结构发现, 在活性位点中正好有一个水分子位于底物Cα=Cβ双键的附近, 而且处于Glu144和Glu164残基的氢键距离之内, 这能有效保证水加成反应的顺利发生.在此之前, Cui等[19]已经通过DFT计算发现Glu144和Glu164都以去质子化的状态存在时ECH催化活性最高.因此, 我们的计算模型直接使用去质子化的Glu144和Glu164.通过DFT计算, 我们建议了一个分步的DAC-CoA水合机理, 如图 4所示.在ECH中, 底物以合适的朝向结合进活性位点, 形成一个稳定的酶-底物复合物(ESDAC-CoA). Glu164残基先作为一个广义碱从水分子中夺取一个质子, 同时水分解形成的活性羟基作为亲核试剂进攻底物的Cβ原子, 形成一个带负电荷的烯醇中间体(EIDAC-CoA).随后, Glu164把从水中得到的质子转移给底物Cα原子, 形成最终的酶-产物复合物(EPDAC-CoA).
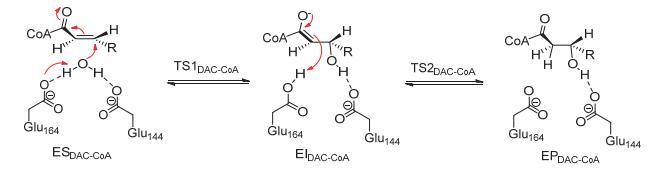 图 4
DFT计算建议的ECH催化DAC-CoA水合的机理
Figure 4.
ECH-catalyzed hydration mechanism of DAC-CoA proposed by DFT calculations
图 4
DFT计算建议的ECH催化DAC-CoA水合的机理
Figure 4.
ECH-catalyzed hydration mechanism of DAC-CoA proposed by DFT calculations
DAC-CoA水加成中所有优化的驻点构型及相应的能量关系如图 5所示.在ESDAC-CoA中, 活性H2O与Glu144和Glu164的羧基以及Gly172的NH基团形成了很强的氢键作用, 距离分别是1.81 Å、1.79 Å和1.78 Å.这些氢键作用把水分子合理定位在底物Cα=Cβ双键的附近, 使其能有效活化底物.此外, 底物羰基与Ala98和Gly141的NH基团形成氢键, 距离分别是1.91 Å和2.33 Å, 这两个氢键的存在能准确定位底物α, β-不饱和硫酯部分的朝向, 使其利于发生水合反应. TS1DAC-CoA对应的是Glu164从H2O中夺取质子同时游离羟基亲核进攻底物Cβ原子的过渡态.在TS1DAC-CoA中, Glu164的羧基O原子与H2O中的H原子之间的距离从1.79 Å缩短到1.41 Å, 同时H2O中O原子与底物Cβ原子之间的距离也从ESDAC-CoA中的3.36 Å骤减到1.81 Å.此外, 亲核反应的发生增强了Gly141的NH基团与底物C=O之间的氢键作用, 氢键距离从ESDAC-CoA中的2.33 Å缩短到2.00 Å, 并进一步缩短到EIDAC-CoA中的1.82 Å.而Ala98的NH基团与C=O之间的氢键距离也缩短到EIDAC-CoA中的1.87 Å.事实上, OH亲核进攻Cβ原子破坏了底物中的共轭大π键, 并在一定程度上增加了羰基O原子的电荷密度.这些结果表明Ala98和Gly141能有效稳定反应中产生的过渡态和烯醇负离子中间体.计算的TS1DAC-CoA的虚频是294.4i cm-1, 能垒是22.2 kcal/mol, 而且EIDAC-CoA的能量要比ESDAC-CoA高9.2 kcal/mol, 这表明该亲核反应是一个不易发生的吸热过程.
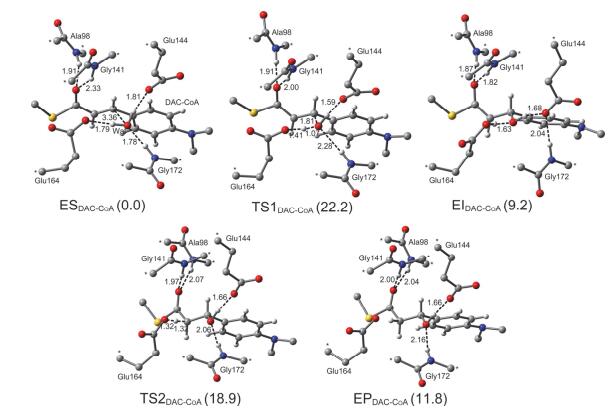 图 5
优化的DAC-CoA水加成路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 5.
Optimized structures of reactant, transition states, intermediates, and product for DAC-CoA hydration pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses
图 5
优化的DAC-CoA水加成路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 5.
Optimized structures of reactant, transition states, intermediates, and product for DAC-CoA hydration pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses
相对于ESDAC-CoA, EIDAC-CoA在能量上很不稳定, 其上的Cα原子需要夺取一个外部质子以确保最终产物EPDAC-CoA的形成, 而ECH活性位点分析及DFT计算表明已经质子化的Glu164是唯一合适的质子供体. TS2DAC-CoA对应于从Glu164到Cα原子的质子转移过渡态.在TS2DAC-CoA中, Cα原子与Glu164中的羧酸H原子之间的距离为1.32 Å, 而Ala98和Gly141与C=O之间的氢键距离分别为2.07 Å和1.97 Å. TS2DAC-CoA的虚频907.6i cm-1.相对于EIDAC-CoA, 计算的TS2DAC-CoA的能垒是9.7 kcal/mol, 远小于TS1DAC-CoA, 这表明该步质子转移反应很容易发生.总的来说, ECH催化的DAC-CoA水加成是一个吸热反应, 整个催化过程需要吸收11.8 kcal/mol的能量.亲核反应的能垒要远高于质子转移(22.2 vs. 9.7 kcal/mol), 是整个催化反应的决速步.需要指出的是, 亲核反应的能垒要比一般的酶催化反应的能垒稍高, 这可能是因为共轭大π键的存在使底物表现出较高的稳定性.从计算的能垒来看, DAC-CoA水加成反应的化学平衡更倾向于反应物, 而不是形成水合产物.因此, DAC-CoA并不是ECH催化的理想底物, 这与实验结果一致[8, 33].
事实上, 我们也尝试探讨了DAC-CoA水加成的协同机理.早期的实验根据动力学同位素效应建议了一个协同的C=C双键水合机理, 其中H2O的分解以及C—OH键和C—H键的形成只需一步基元反应, 不会涉及到烯醇负离子中间体[3, 17].然而, 我们并没有表征出合理的“四元环”协同过渡态, 只能得到分步机理中涉及到的两个过渡态.理论上来说, 四元环过渡态的环张力较大, 使其在能量上很不稳定, 而且也不易被准确优化.因此, DAC-CoA水加成应该以分步机理而不是协同机理进行, 这与先前的理论计算结果保持一致[18, 19].
3.2 Crotonyl-CoA水合反应
我们进一步研究了ECH催化的Crotonyl-CoA水合反应.该反应与DAC-CoA水加成类似, 以分步机理进行, 经历一个烯醇负离子中间体.为了与DAC-CoA水合反应对比, 我们先采用相同的活性位点模型研究Crotonyl-CoA水合反应, 标记为模型A, 相应的反应物为A-ESCr-CoA.优化的模型A路径中所有的驻点构型及相应的能量关系如图 6所示.在A-ESCr-CoA中, Ala98和Gly141与底物C=O之间的氢键距离分别为1.83 Å和2.19 Å, 氢键作用要稍强于这两个氨基酸与DAC-CoA底物C=O之间的作用.上面已经提到, DAC-CoA中的芳环与α, β-不饱和硫酯部分之间较强的共轭作用增加了底物的稳定性.而在Crotonyl-CoA中, 甲基取代了芳环, 降低了底物的稳定性, 相反也就增加了底物的水加成活性.模型A中亲核反应的能垒是18.7 kcal/mol, 质子转移的能垒为5.1 kcal/mol, 整个催化过程需要吸热6.6 kcal/mol, 反应所需的能量明显低于DAC-CoA水加成, 这些结果表明Crotonyl-CoA要比DAC-CoA更易被H2O活化.需要指出的是, 实验上并没有对DAC-CoA和Crotonyl-CoA水加成是吸热或者放热反应进行相关报道, 或许通过实验手段直接测定酶催化反应的吸放热量并不容易(尤其在吸放热量并不明显的情况下), 但是我们的计算结果显示这两个水加成反应都是吸热的, 这表明理论计算在研究酶催化反应方面有独特优势.理论上, α, β-不饱和硫酯底物中由于共轭π键的存在, 其稳定性应该要高于水合产物, 因此ECH水合反应应该是吸热的.
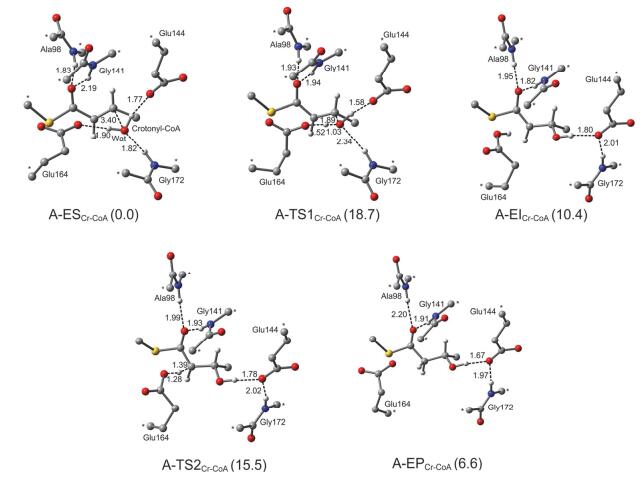 图 6
优化的Crotonyl-CoA水加成A路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 6.
Optimized structures of reactant, transition states, intermediates, and product for Crotonyl-CoA hydration A-pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses
图 6
优化的Crotonyl-CoA水加成A路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 6.
Optimized structures of reactant, transition states, intermediates, and product for Crotonyl-CoA hydration A-pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses
随后, 为准确探讨Crotonyl-CoA水合活性, 我们在模型A的基础上又构建了一个更大的模型, 标记为B, 包括模型A中的所有原子、Gln162以及Glu164、Ala173和Gly175的骨架酰胺基团, 共115个原子. 图 7列出了优化的模型B路径中所有的驻点构型及相应的能量关系.在B-ESCr-CoA中, Glu164的骨架N—H与其侧链羧基形成氢键, 距离为1.81 Å.而Glu144的羧基也与Ala173和Gly175的骨架N—H形成氢键, 距离分别是1.85 Å和1.88 Å.这些氢键作用能有效稳定活性位点的排布.把这些氢键作用加入到计算模型中既可以在一定程度上消除构型优化过程中Glu144和Glu164残基侧链朝向的不合理变化所带来的附加误差, 也能更好地考虑酶环境对反应的影响.事实上, 优化的B-ESCr-CoA构型能与晶体结构活性位点更好地吻合, 这说明我们构建的该计算模型是合理且可靠的.在B路径中, 计算的亲核反应的能垒是12.9 kcal/mol, 明显小于模型A中的能垒.意外的是, 质子转移的能垒是6.1 kcal/mol, 稍高于模型A.对比优化的A-ESCr-CoA和A-EICr-CoA构型发现, 亲核反应的发生使Gly172自发地破坏了其与H2O之间的氢键作用, 转而与Glu144的羧基形成氢键.这种变化很不正常, 因为在真实的酶三维立体结构中肽键的朝向一般不会发生很大变化.此外, 由于缺少氢键网络的限制, Glu144和Glu164残基侧链的朝向随着反应的发生变化也很明显.这些结果说明模型A并不是很合理.相反, 模型B真实地还原了ECH活性位点的部分氢键网络结构, 水合反应的发生并没有破坏掉Gly172与H2O之间的氢键作用, 而且Glu144和Glu164残基侧链的朝向也没有发生不合理偏转.此外, 模型B中氢键网络的存在使得活性水分子与底物Cα=Cβ双键之间能保持比较理想的相对朝向, 这更有利于亲核反应的发生, 进而使得其亲核反应的能垒(12.9 kcal/mol)明显小于模型A (18.7 kcal/mol).因此, 构建的模型B要比模型A更合理, 而且模型B中的能量数据更能准确反映ECH的催化活性.
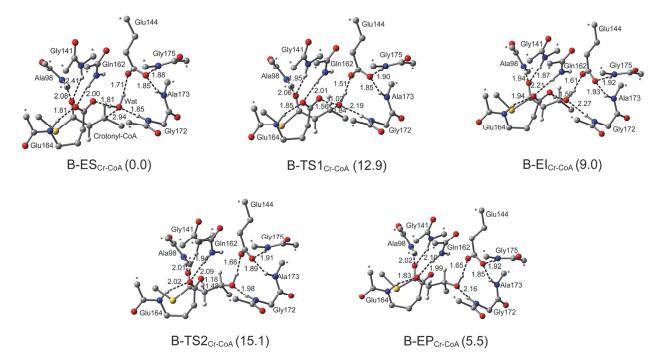 图 7
优化的Crotonyl-CoA水加成B路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 7.
Optimized structures of reactant, transition states, intermediates, and product for Crotonyl-CoA hydration B-pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses
图 7
优化的Crotonyl-CoA水加成B路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 7.
Optimized structures of reactant, transition states, intermediates, and product for Crotonyl-CoA hydration B-pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses
ECH不仅能催化DAC-CoA和Crotonyl-CoA发生水合反应, 还能催化其他多种α, β-不饱和硫酯底物水合, 如3′-脱磷酸巴豆酰-辅酶A (dpCr-CoA)、(2-苯并噻唑)-4-硫代丁酰基-辅酶A (BTTB-CoA)、己二烯酰基-辅酶A (HD-CoA)和肉桂酰基-辅酶A (Cin-CoA)等[13, 16].这些研究表明ECH对多种不同取代基的烯酰基-辅酶A底物都有比较好的催化活性.基于此, 在有机合成上可以广泛应用ECH催化同类型的α, β-不饱和硫酯化合物水合, 其催化效率高且选择性好.
3.3 活性位点氨基酸作用
实验研究已经明确指出, Ala98和Gly141能有效促进α, β-不饱和硫酯的水加成反应[8, 15]. Gly141在ECH家族中是一个保守的残基, 把Gly141突变成Pro会导致ECH催化活性急剧降低[12, 15].尽管Ala98并不是一个保守残基, 但是把Ala98突变成Pro也会显著降低酶催化活性[12].活性位点结构分析和DFT计算表明, Ala98和Gly141的骨架N—H能与α, β-不饱和硫酯的C=O形成氢键作用.一方面, 这两个氢键可以诱使底物α, β-不饱和硫酯部分结合进ECH活性位点, 并使其保持一个合理的朝向以利于催化水分子亲核进攻Cα=Cβ双键.另一方面, 这两个氢键可以通过形成一个“氧负离子穴”极化底物, 促进水合反应的发生, 进而稳定反应产生的过渡态和烯醇负离子中间体.定点突变实验研究表明, 在ECH催化的Crotonyl-CoA水解反应中, Glu164Gln突变体的催化活性几乎完全丧失(催化速率常数kcat降低约3×105倍)[13], 这说明Glu164直接参与底物的水合, 与我们的计算结果相一致.尽管Glu144没有参与反应, 但是它能通过与H2O之间的强氢键作用使H2O中的O原子以合适的朝向亲核进攻底物Cβ原子, 而且突变试验也已经证实了它的重要性.同样, Gly172与H2O之间的氢键作用对于底物活化也很重要.此外, Glu164、Ala173和Gly175的骨架N—H及Gln162侧链与Glu164和Glu144之间的氢键网络对于维持活性位点的有效排布进而促进底物活化也是必不可少的.
4 结论
本文使用DFT方法研究了ECH催化的DAC-CoA和Crotonyl-CoA水合反应.计算结果表明水合反应以E1cb机理进行: Glu164残基先作为催化碱从水分子中夺取一个质子, 同时产生的游离羟基亲核进攻底物Cβ原子, 形成一个能量上不稳定的烯醇负离子中间体; 随后, 已经质子化的Glu164作为酸提供一个质子给中间体的Cα原子, 得到最终的水合产物. DAC-CoA底物中的芳环和α, β-不饱和硫酯部分能形成较强的共轭作用, 使其表现出较高的稳定性, 这在一定程度上增加了水合反应的难度, 也诱使反应的化学平衡更倾向于ECH-DAC-CoA复合物.而Crotonyl-CoA中由于没有芳环, 它的水加成活性要显著高于DAC-CoA.此外, 研究结果表明DAC-CoA和Crotonyl-CoA水加成都是吸热的反应. Ala98和Gly141可以与底物α, β-不饱和硫酯部分的C=O形成氢键, 它们的存在既有利于底物在ECH活性位点的准确结合, 也能有效稳定反应中产生的过渡态和中间体. Glu144残基虽然没有直接参与水加成反应, 但是它能通过与水分子形成氢键诱使水分子以合理的朝向参与反应.另外, Glu144和Glu164周围的氢键网络能合理维持ECH活性位点的排布, 进而有效促进底物活化.
-
-
[1]
Willadsen, P.; Eggerer, H. Eur. J. Biochem. 1975, 54, 247. doi: 10.1111/ejb.1975.54.issue-1
-
[2]
Wu, W. J.; Feng, Y.; He, X.; Hofstein, H. A.; Raleigh, D. P.; Tonge, P. J. J. Am. Chem. Soc. 2000, 122, 3987. doi: 10.1021/ja992286h
-
[3]
Bahnson, B. J.; Anderson, V. E. Biochemistry 1989, 28, 4173. doi: 10.1021/bi00436a008
-
[4]
Müller-Newen, G.; Janssen, U.; Stoffel, W. Eur. J. Biochem. 1995, 228, 68. doi: 10.1111/ejb.1995.228.issue-1
-
[5]
Boersma, A. J.; Coquière, D.; Geerdink, D.; Rosati, F.; Feringa, B. L.; Roelfes, G. Nat. Chem. 2010, 2, 991. doi: 10.1038/nchem.819
-
[6]
Silverman, R. B. The Organic Chemistry of Enzyme-catalyzed Reactions, Academic, London, 2002, pp. 428~448.
-
[7]
Engel, C. K.; Mathieu, M.; Zeelen, J. P.; Hiltunen, J. K.; Wierenga, R. K. EMBO J. 1996, 15, 5135.
-
[8]
Bahnson, B. J.; Anderson, V. E.; Petsko, G. A. Biochemistry 2002, 41, 2621. doi: 10.1021/bi015844p
-
[9]
Hisano, T.; Tsuge, T.; Fukui, T.; Iwata, T.; Miki, K.; Doi, Y. J. Biol. Chem. 2003, 278, 617.
-
[10]
Koski, M. K.; Haapalainen, A. M.; Hiltunen, J. K.; Glumoff, T. J. Biol. Chem. 2004, 279, 24666. doi: 10.1074/jbc.M400293200
-
[11]
Baugh, L.; Phan, I.; Begley, D. W.; Clifton, M. C.; Armour, B.; Dranow, D. M.; Taylor, B. M.; Muruthi, M. M.; Abendroth, J.; Fairman, J. W.; Fox, D. 3rd; Dieterich, S. H.; Staker, B. L.; Gardberg, A. S.; Choi, R.; Hewitt, S. N.; Napuli, A. J.; Myers, J.; Barrett, L. K.; Zhang, Y.; Ferrell, M.; Mundt, E.; Thompkins, K.; Tran, N.; Lyons-Abbott, S.; Abramov, A.; Sekar, A.; Serbzhinskiy, D, ; Lorimer, D.; Buchko, G. W.; Stacy, R.; Stewart, L. J.; Edwards, T. E.; Van Voorhis, W. C.; Myler, P. J. Tuberculosis 2015, 95, 142.
-
[12]
Feng, Y.; Hofstein, H. A.; Zwahlen, J.; Tonge, P. J. Biochemistry 2002, 41, 12883. doi: 10.1021/bi020382g
-
[13]
Hofstein, H. A.; Feng, Y.; Anderson, V. E.; Tonge, P. J. Biochemistry 1999, 38, 9508. doi: 10.1021/bi990506y
-
[14]
Engel, C. K.; Kiema, T. R.; Hiltunen, J. K.; Wierenga, R. K. J. Mol. Biol. 1998, 275, 847. doi: 10.1006/jmbi.1997.1491
-
[15]
Bell, A. F.; Wu, J.; Feng, Y.; Tonge, P. J. Biochemistry 2001, 40, 1725. doi: 10.1021/bi001733z
-
[16]
D'Ordine, R. L.; Pawlak, J.; Bahnson, B. J.; Anderson, V. E.; Biochemistry 2002, 41, 2630. doi: 10.1021/bi015845h
-
[17]
Bahnson, B. J.; Anderson, V. E. Biochemistry 1991, 30, 5894. doi: 10.1021/bi00238a013
-
[18]
Pawlak, J.; Bahnson, B.; Anderson, V. Nukleonika 2002, 47, 33.
-
[19]
Cui, X.; He, R.; Yang, Q.; Shen, W.; Li, M. J. Mol. Model. 2014, 20, 2411. doi: 10.1007/s00894-014-2411-5
-
[20]
Agnihotri, G.; Liu, H. W. Bioorg. Med. Chem. 2003, 11, 9. doi: 10.1016/S0968-0896(02)00333-4
-
[21]
Siegbahn, P. E.; Himo, F. J. Biol. Inorg. Chem. 2009, 14, 643. doi: 10.1007/s00775-009-0511-y
-
[22]
Siegbahn, P. E.; Blomberg, M. R. Chem. Rev. 2010, 110, 7040. doi: 10.1021/cr100070p
-
[23]
Hopmann, K. H.; Himo, F. In Comprehensive Natural Products Chemistry Ⅱ Chemistry and Biology, Vol. 8, Eds.: Mander, L. N.; Liu, H.-W., Elsevier, Oxford, 2010, pp. 719~747.
-
[24]
Siegbahn, P. E.; Himo, F. Wiley Interdisciplinary Reviews: Computational Molecular Science, 2011, 1, 323. doi: 10.1002/wcms.13
-
[25]
Blomberg, M. R.; Borowski, T.; Himo, F.; Liao, R. Z.; Siegbahn, P. E. Chem. Rev. 2014, 114, 3601. doi: 10.1021/cr400388t
-
[26]
Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/1.464913
-
[27]
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785. doi: 10.1103/PhysRevB.37.785
-
[28]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03 (Revision D. 01), Gaussian, Inc., Wallingford CT, 2004.
-
[29]
Barone, V.; Cossi, M.; Tomasi, J. J. Comput. Chem. 1998, 19, 404. doi: 10.1002/(ISSN)1096-987X
-
[30]
Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027. doi: 10.1021/cr00031a013
-
[31]
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi: 10.1063/1.3382344
-
[32]
Grimme, S.; Ehrlich, S.; Goerigk, L. J. Comput. Chem. 2011, 32, 1456. doi: 10.1002/jcc.v32.7
-
[33]
D'Ordine, R. L.; Tonge, P. J.; Carey, P. R.; Anderson, V. E. Biochemistry 1994, 33, 12635. doi: 10.1021/bi00208a014
-
[1]
-

图 1 ECH催化的α, β-不饱和硫酯水合反应
Figure 1 Hydration of α, β-unsaturated thiolester catalyzed by ECH


图 3 DFT计算构建的活性位点模型示意图.底物中R可以是4-(N, N-二甲氨基)-苯基或者甲基.红色虚线代表可能的氢键作用
Figure 3 Constructed active site model for DFT calculations. R group in substrate can be 4-dimethylamino-phenyl or methyl. Red dashed lines represent the possible hydrogen-bonding interactions

图 4 DFT计算建议的ECH催化DAC-CoA水合的机理
Figure 4 ECH-catalyzed hydration mechanism of DAC-CoA proposed by DFT calculations

图 5 优化的DAC-CoA水加成路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 5 Optimized structures of reactant, transition states, intermediates, and product for DAC-CoA hydration pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses

图 6 优化的Crotonyl-CoA水加成A路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 6 Optimized structures of reactant, transition states, intermediates, and product for Crotonyl-CoA hydration A-pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses

图 7 优化的Crotonyl-CoA水加成B路径中反应物、过渡态、中间体和产物的构型, 距离单位为Å.相应的能量数据在括号中给出, 单位为kcal/mol
Figure 7 Optimized structures of reactant, transition states, intermediates, and product for Crotonyl-CoA hydration B-pathway. All distances are shown in angstroms. Relative energies (kcal/mol) are given in parentheses
-


 下载:
下载:






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 28
- 文章访问数: 3114
- HTML全文浏览量: 984
 下载:
下载:
