Figure 1.
Structure diagrams of complexes 1~4 and FL
Probing Photocatalytic Hydrogen Evolution of Cobalt Complexes: Experimental and Theoretical Methods
Hai-Su WU , Ti-Fang MIAO , Hai-Xia SHI , Yun XU , Xian-Liang FU , Li QIAN

Hydrogen (H2), a source of clean and renewable fuel, has attracted great interest for its potential to store solar energy and to reduce the current dependence on fossil fuels[1, 2]. Reduction of water to H2 with visible light as trigger force has been a subject of attractive attention, and great efforts have been devoted to the design of metal complexes for proton reduction[3-5]. Over the past decades, for reasons of price and practicality, a considerable number of photocatalysts for H2 generation based on non-precious metal complexes such as Fe, Co, Ni, Cu, and Mn have been reported[6-16]. Recently, Li and co-workers have described the photocatalytic proton reduction catalyzed by a cobalt(II)-salen complex with a remarkable turnover number (TON) of > 319 in a MeOH-H2O solution[17]. Although there has been significant progress in designing molecular catalysts for H2 evolution, the search of efficient, robust, and earth-abundant metal photocatalysts that can work in aqueous solution still require further development.
With good performance and low cost, Co-complexes supported by different organic ligands have been reported to catalyze the reduction or oxidation of water in organic solution[3, 18-26]. However, the hydrogen production performance in pure water was hardly reported. Moreover, the current hydrogen-evolution mechanism for Co complexes is still vague, adding difficulty for us to design catalysts and to select photosensitizers. To find out the mechanism of hydrogen released, Co(Ⅲ) complexes with Schiff base were synthesized, and their structure diagrams are shown in Fig. 1. Compared with the reported hydrogen-evolution system, our system was tested in aqueous solution, which was composed of Co complex, fluorescein (FL) and triethanolamine (TEOA) as a catalyst, a photosensitizer and an electron donor, respectively. Here, Co-complexes provided active sites by obtaining electrons from FL under light irradiation, facilitating the proton reduction. Moreover, the best catalytic environment was found by changing the pH and the concentration of photosensitizer continually. Compared with the experimental method, density functional theory (DFT) provides us a convenient way to understand the relationship between structure and properties, and helps us to explore the hydrogen-evolution mechanism of Co complexes[27, 28]. In this work, the interaction properties between photosensitizers and catalysts were calculated, and the hydrogen-evolution reason of Co complexes was explored. The results will be favorable for us to understand the hydrogen-evolution mechanism, design the catalyst and select the photosensitizer.
Cobalt(II) acetate tetrahydrate (C4H6CoO4·4H2O), salicylaldehyde (C7H6O2), ethylenediamine (N2H4), 1, 2-diaminocyclohexane (C6H14N2), 2, 3-diaminopyridine (C5H7N3), o-phenylenediamine (C6H8N2), ethanol, dichloromethane (CH2Cl2), acetic acid, acetonitrile (MeCN), fluorescein (FL), and triethylamine (TEOA) were purchased from Macklin Reagent (Shanghai, China) and used without any further purification.
The ligands were synthesized according to reported literatures modified slightly[29-34]. A solution of salicylaldehyde (4 mmol) and methanol (15 mL) was added dropwise to a solution of ethylenediamine (2 mmol) and methanol (10 mL). The obtained mixture was refluxed for 2 h, which caused a precipitate to form. Then, the precipitate was separated by filtration, washed with cold methanol and dried in vacuum. Finally, it was recrystallized from dichloro-methane to yield as a pure Schiff base ligand.
(R, R)-N, N΄-bis-salicylidene-1, 2-cyclohexanediamine Golden yellow solid, yield 0.264 g, 82%; m.p. 115~117 ℃. 1H NMR (600 MHz, d6-CDCl3): δ 13.34 (s, 2H), 8.28 (s, 2H), 7.26 (t, J = 7.8 Hz, 2H), 7.17 (dd, J = 7.9, 1.8 Hz, 2H), 6.91 (d, J = 8.3 Hz, 2H), 6.81 (t, J = 7.4 Hz, 2H), 3.36~3.31 (m, 2H), 1.99~1.94 (m, 2H), 1.92~1.87 (m, 2H), 1.78~1.72 (m, 2H), 1.52~1.48 (m, 2H). 13C NMR (600 MHz, d6-CDCl3): δ 164.77, 164.21, 161.31, 161.03, 132.30, 132.22, 131.55, 131.52, 118.90, 118.72, 118.65, 118.57, 117.07, 116.82, 72.67, 69.50, 33.15, 30.75, 24.23, 22.55. HRMS (ESI): calcd. for [C20H18N2O2+H]+ 323.1754, found 323.1756.
(R, R)-N, N΄-bis-salicylidene-o-phenylenediamine Yellow solid, yield 0.269 g, 85%; m.p. 165~166 ℃. 1H NMR (600 MHz, d6-DMSO): δ 12.97 (s, 2H), 8.92 (s, 2H), 7.66 (d, J = 7.7 Hz, 2H), 7.50~7.36 (m, 6H), 6.96 (d, J = 7.9 Hz, 4H). 13C NMR (600 MHz, d6-DMSO) δ 164.05, 160.42, 142.27, 133.47, 132.50, 127.84, 119.76, 119.50, 119.11, 116.70. HRMS (ESI): calcd. for [C20H18N2O2+H]+ 317.1285, found 317.1286.
(R, R)-N, N΄-bis-salicylidene-2, 3-diaminopyridine
Yellow solid, yield 0.257 g, 80%; 1H NMR (600 MHz, d6-DMSO): δ 12.88 (d, J = 310.0 Hz, 2H), 9.56 (s, 1H), 8.98 (s, 1H), 8.49~8.41 (m, 1H), 7.92 (dd, J = 7.8, 1.5 Hz, 1H), 7.82~7.78 (m, 1H), 7.71 (dd, J = 7.6, 1.7 Hz, 1H), 7.51~7.43 (m, 3H), 7.04~6.96 (m, 4H). 13C NMR (600 MHz, d6-DMSO): δ 165.52, 164.15, 161.12, 160.29, 150.57, 146.57, 139.07, 134.21, 133.81, 133.15, 132.22, 128.98, 123.92, 119.55, 119.23, 116.78, 116.73. HRMS (ESI): calcd. for [C19H16N3O2+H]+ 318.1237, found 318.1268.
Co(II)-salen complexes were synthesized from salen ligands and cobalt(II) acetate in ethanol according to the procedure described[35-37]. Co(III)-salen complexes were then purified according to the reported literatures[37, 38]. That is, 0.45 mL acetic acid was added to the Co(II)-salen complex (0.20 g) in dichloromethane (12 mL). After stirring the mixture for 50 min, the solvent of dichloromethane was removed with a rotavapor and the excess acetic acid was also removed under vacuum[39-44]. Since the complexes were too insoluble to allow solution NMR and polarimetry studies, their infrared spectrum data were measured.
[N, N΄-bis(salicylidene)-trans-1, 2-cyclohexanediamine] cobalt(II). Red solid, yield 62%, m.p. 300~302 ℃; IR (KBr, cm-1) v(C=N) 1603, vs(Co=O) 627, v(Co=N) 415; HRMS-ESI m/z[Co]+ calcd. for C20H20CoN2O2: 379.0857, found: 379.0851.
[N, N΄-bis(salicylidene)-o-phenylenediamine] cobalt(II) Brown solid, yield 64%, m.p. > 300 ℃; IR (KBr, cm-1) v(C=N) 1610; HRMS-ESI m/z[Co]+ calcd. for C20H14CoN2O2: 373.0387, found: 373.0383.
[N, N΄-bis(salicylidene)-2, 3-diaminopyridyl] cobalt(II) Brown solid, yield 58%, m.p. > 300 ℃; IR (KBr, cm-1) v(C=N) 1615, v(Co=N) 550, v(Co=O) 468; HRMS-ESI m/z[Co]+ calcd for C20H14CoN2O2: 374.0340, found: 374.0346.
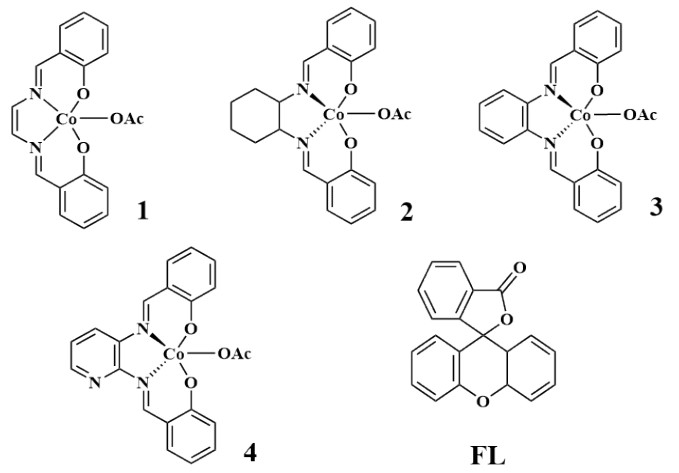
The peak around 3030 cm-1 was assumed to be the C–H stretching vibration on the phenyl group (Fig. 2). There is no peak around 2560 cm-1. It is believed that the ligand and the central metal are coordinated together[45]. The four complexes have strong vibration peaks around 1290 cm-1, which is caused by the stretching vibration of the C–O bond. Similar ligands show peaks[46-48] in the range of 1280~1340 cm-1. The stretching vibration of C=N bond has moved to a lower frequency[49, 50]. Since the C=N is coordinated with the central metal Co, Vas (Co–O) and Vs (Co–O) can be detected in all complexes. Vas (Co–O) appears around 750 cm-1, and Vs (Co–O) peaks around 650 cm-1. The peaks at 1710 cm-1 are assigned to the carbonyl peak of carboxylate. It can be inferred that the last carboxyl group has been formed by the product of the previous step. The peak at 1540 cm-1 is from the phenyl breathing mode, and that at 415 cm-1 is assigned to the Co–N stretching vibration[50, 51].
Proton NMR spectra were recorded on a Bruker DRX 600 spectrophotometer in deuterated reagent by the residual solvent signal as an internal calibration. High resolution mass spectroscopic (HRMS) data of the products were collected on an Agilent Technologies 6540 UHD Accurate-Mass Q-TOF LC/MS using ESI. Hitachi UV-3600 UV-vis spectra were used to measure the UV-vis spectra with a 1 cm quartz cell. Photoluminescence (PL) emission spectra were measured on a fluorescence spectrometer (JASCO FP-6500) at 20 ℃. FT-IR spectra were recorded on a NEXUS470 spectrophotometer, and KBr was used as the substrate. Cyclic voltammetry (CV) was measured in an electrochemical workstation equipped with a Pt wire as the counter electrode, a glassy carbon as the working electrode, and a Ag/AgCl as the reference electrode at a scan rate of 100 mV/s in 0.1 M Bu4NPF6 DMF solution. The pH value was determined by a PHS-25 pH meter.
Photocatalytic HER was tested in a commercial reaction cell (Lab Solar II, Perfect Light Co., T = 10 ℃). The generated H2 was measured by an online gas chromatograph (GC7900, TCD as detector, Ar as carrier gas). A Xe lamp equipped with a cut-off filter (PLS-SEX 300, λ ≥ 400 nm, 300 W) was used as the light source. The visible light intensity was about 100 mW/cm2 measured by a PL-MW2000 spectroradiometer. In a typical run, a three-component system consisting of 10 mg cobalt-salen molecules (as photocatalyst), a certain amount of FL (as a photosensitizer), and 10 mL TEOA (as sacrificial reagent) was subjected to H2 evolution experiments in 90 mL H2O without another organic reagent added. The pH of the system was adjusted by the addition of HCl and NaOH solutions. The mixed system was stirred in the dark for 30 min before turning on the lamp. During the HER test, the evolved H2 was measured every 30 min by online GC. The apparent quantum efficiency (AQE) of H2 evolution was measured with a band pass filter (λ = 400 nm) by equation (1). The irradiation area was about 3.14 cm2 and the intensity of the irradiation was about 18.8 mW/cm2.
|
|
(1) |
Our previous research[52] shows that the solvent effect is very important for the calculation of cobalt complexes. Hence, the ground-state complexes 1~4 were subjected to full geometry optimization at the B3LYP/6-31G(d) level in aqueous solution by the CPCM model[53, 54]. Optimized minima were verified with vibrational calculations. To study the interaction of Co complexes with FL, the FL was also optimized in aqueous solution by the CPCM model at the same level of theory, and its structural diagram is also shown in Fig. 1.
The optimized complexes were bound to FL by the Dock6.0 program[55, 56]. The box size, grid space, energy cut of distance, and maximum orientation were set as 30, 0.3, 9999, and 150, 000, respectively. Other binding parameters were set to default values, and thus we obtained binding geometries of the complexes with FL, which were further optimized in aqueous solution by the same method. To study the excited properties of the binding geometries, the lowest singlet states of binding geometries were optimized in aqueous solution at the same level of theory. Meanwhile, the optimized minima were verified with the vibrational calculations.
The non-adiabatic electron‑transfer (ET) process is usually tackled by the theory of Marcus semi-classical model[57, 58]. Hence, we can obtain the rate constant by the following equation:
|
|
(2) |
Here, λ is the reorganization energy, h the Planck's constant, HAD the ET matrix element, T the temperature, and ∆G◦ the normal Gibbs free energy change of the ET reaction.
Cobalt complexes binding to FL can be excited by the light irradiation, leading to ET from FL to complex at a moving speed of ket. Take Co-complex and FL as an example, this process can be expressed as follows:
|
|
(3) |
After electron transfer, λ can be computed by the following equation.
|
|
(4) |
Here, λ1 is the reorganization energy of FL and λ2 is the reorganization energy of cobalt complex.
To obtain the accurate reorganization energy, we replaced the Co-complex and FL in reaction (3) with the complex and FL in the optimized binding-geometry in the ground state (S0), respectively. Similarly, we replaced the Co-complex- and FL+ in reaction (3) with the complex and FL in the optimized binding-geometry in the lowest singlet state (S1), respectively. We first obtained the total energy of E(FL) at its geometry in the optimized binding-geometry in S0, and the total energy of E(FL+) at its geometry in the optimized binding-geometry in the S1. Besides, we also calculated the energy E′(FL) at the geometry of FL+ and the energy E′(FL+) at the geometry of FL. Thus, we obtained λ1 by
|
|
(5) |
In the same way, we obtained λ2 by
|
|
The total reorganization energy was calculated via Eqs. 4, 5 and 6.
The ET matrix element HDA is usually estimated by the generalized Mulliken-Hush (GMH) theory, which can be calculated by the following equation[59]:
|
|
(7) |
where ΔEij is the energy gap between the ground state and the excited one, Δuij is the dipole moment difference between states i and j, and mij denotes the transition dipole moment connecting the two states, which were the optimized binding-geometries in S0 and S1.
The binding-geometry of cobalt complex with FL can be excited by the light irradiation. The process can be expressed as follows:
|
|
(8) |
Where Co-complex-FL and ′Co-complex-FL indicate the binding-geometries in S0 and S1, respectively. The standard Gibbs free energy change of the ET reaction (8) was calculated by:
|
|
(9) |
Where GCo-complex-FL and G′Co-complex-FL are the energies of the optimized binding-geometries in S0 and S1, respectively. Meanwhile, the correction to Gibbs free energy was added to G and G′.
The calculations above were performed with Gaussian16 program-package[60].
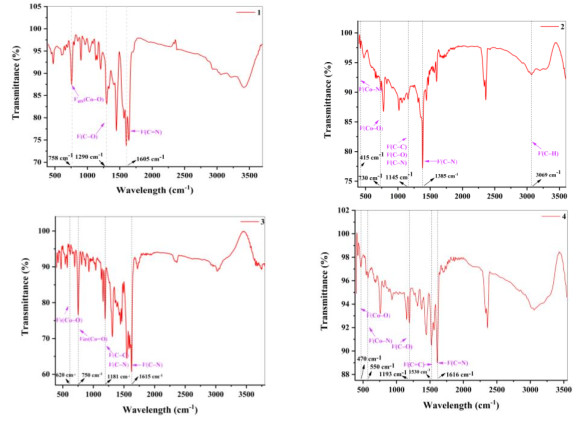
The UV-vis absorption spectra of complexes 1~4 were measured at room temperature in MeCN: H2O = 1:1 (see Fig. 3). All complexes exhibited a strong-energy peak at about 260 nm and a poor light absorption in the visible region. Interestingly, complexes 3 and 4 have weaker absorption peaks near 325 and 330 nm, respectively, whereas 1 and 2 have not. Besides, the FL demonstrated a strong visible light absorption observed at about 480 nm, which ensured the possibility to trigger the visible-light-induced water splitting.
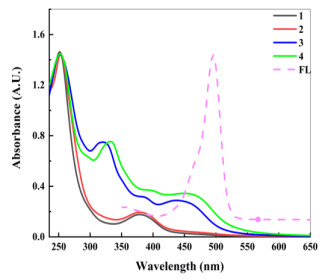
The PL spectra of complexes 1~4 were measured and shown in Fig. 4. Generally speaking, weaker fluorescence signals show weaker photogenerated carrier recombination[61, 62]. According to the viewpoint, complex 3 should have strong ability to promote the separation of photogenerated charge carriers relative to complexes 1 and 2. This suggests that the recombination of photogenerated charge carriers can be delayed effectively for 3, resulting in a lot of H2 generated. Although the fluorescence performance of 4 was weaker than that of 3 in Fig. 4, the hydrogen-evolution amount of 4 was less than that of 3. The reason may be that the N atom in the pyridine ring on the equatorial plane inhibits the separation of photogenerated charge carriers and the performance of hydrogen production. In general, the peak shapes of PL spectra for the four complexes were similar, and the photocatalytic performances of hydrogen production were more or less the same (see the following 'Photocatalytic H2 production' section). This indicates that modifying different groups for Schiff base complexes have a little impact on the hydrogen-production performance[63, 64].
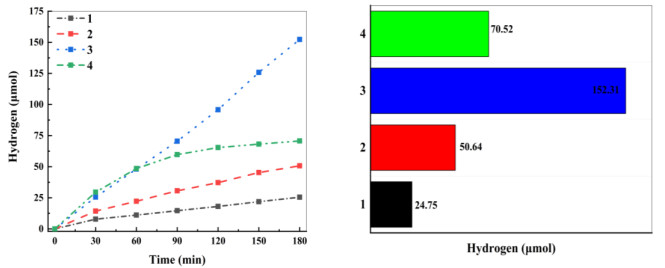
The photocatalytic performance of complexes for hydrogen production was evaluated. The time-course of H2 amount is shown in Fig. 5. It indicates that, after irradiation for 180 min, about 24.07, 48.24, 152.3, and 63.73 μmol of hydrogen are released over complexes 1, 2, 3, and 4, respectively. Obviously, the catalytic hydrogen-production performance of complex 3 is significantly better than that of the other complexes in aqueous solution. It also indicates that the hydrogen-production performance of complex 3 in aqueous solution is better than that of complex 1. In addition, the hydrogen-production performance of complex 1 was also tested in the mixed solution of ethanol and water as a try, showing that the H2 amount in the mixed solution is less than that in aqueous solution. Such a result gives us a little insight, i.e., the hydrogen-production performance of Co complexes can be improved by substituting the aqueous solution for the organic solvent. Meanwhile, this practice can effectively reduce the environmental pollution and the cost.
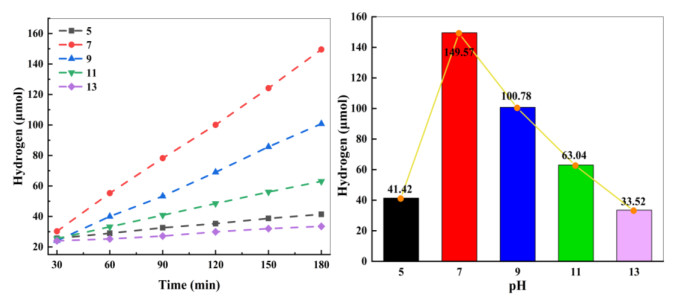
The photosensitizer concentration and the pH of medium affect the HER performance, so they were optimized with complex 3 as the catalyst. As shown in Fig. 6, when the FL amount is less than 12 mg, the rH2 of complex 3 is almost proportional to the FL amount. Further increasing the amount of FL to 14 mg can rapidly reduce rH2 from a maximum of 152.3 to 92.30 μmol, indicating that the ratio of PS to catalyst is a key factor to influence the evolution of hydrogen in the system, which may be due to the saturation of electrons provided by FL when the concentration was too high. The presence of sufficient FL greatly increases the chances of the catalyst obtaining electrons, thereby improving the catalytic efficiency of the system. However, excessive FL is detrimental to its dispersion and the absorption of visible light, thus decreasing the H2 amount. An analogous phenomenon was reported in the literature[65].

Moreover, the pH can significantly affect the interaction among photosensitizer, catalyst and TEOA, and further affects the evolution of H2 in the photocatalytic system[66]. Fig. 7 illustrates the variation of H2 amount with the initial pH value in the range from 5 to 13. The maximal amount of H2 (ca. 152.3 μmol) can be obtained at pH = 7, whereas the hydrogen production drops sharply when the pH of the system is greater or less than 7. Fig. 7 also shows that the amount of hydrogen was only 23.52 μmol after three hours of light at pH = 13. When pH = 5, the detected amount of hydrogen is 31.42 μmol. When the alkalinity is too high, the proton concentration in the system is significantly reduced, making difficult protonation for the intermediates, which affects proton reduction and the evolution of H2. Strongly acidic solution can cause TEOA protons, resulting in a poor supply capacity. This may be the main reason of the decrease in hydrogen generation under lower pH.
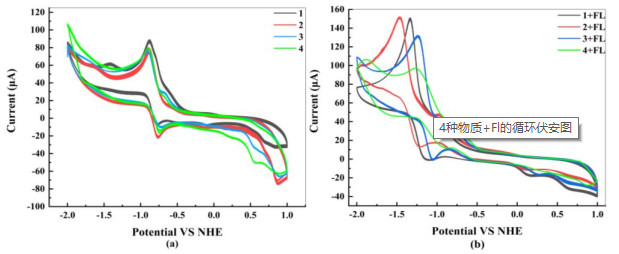
The cyclic voltammograms of complexes were tested in a 0.1 M Bu4NPF6 DMF solution. The redox potentials were obtained through the Ag/AgCl pair, and were adjusted to a standard hydrogen electrode. As shown in Fig. 8a, the reversible redox potentials are at −0.625, −0.602, −0.661 and −0.670 V (vs NHE), corresponding to the CoIII/CoII redox couples of complexes 1~4. Moreover, complexes 1~4 display redox potentials at −0.785, −0.762, −0.735 and −0.754 V (vs NHE), respectively, tentatively assigned as the CoII/CoI couple. As shown in Fig. 8b, after the addition of FL, there is a significant cathodic shift for each catalyst. The CoIII/CoII redox couples of complexes 1~4 change into −0.954, −0.967, −0.862 and −0.811 V. Redox potentials of complexes 1~4 are at −1.130, −1.223, −1.055 and −0.912 V, respectively, corresponding to the redox pairs of CoII/CoI. This is mainly attributed to the electron donation of FL. Such a result indicates that the main influence on the catalytic performance of complexes is not the potential, but effective ET and photogenerated carrier recombination.
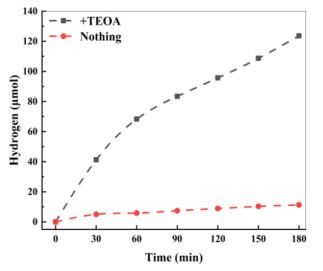
The essence of photocatalytic hydrogen production is that electrons in the photosensitizer are excited creating photogenerated electrons, which are quickly transferred to the active site of the catalyst. The rapid ET makes the concentration of electrons on the active site of the catalyst expand rapidly, and the hydrogen protons in the solution get these electrons easily, leading to hydrogen gas generated. Research finds that many catalysts usually cause the photo-generated electrons to recombine due to some defects. Fortunately, the electronic sacrificial agent can suppress the recombination, improve the quantum conversion efficiency and increase the hydrogen-evolution amount. As shown in Fig. 9, under optimal conditions, the hydrogen production results of complex 3 were studied with or without electron sacrificial agent. We can see that, after 3 h of irradiation, the hydrogen-evolution amount with the electron sacrificial agent was 38 times that without the electron sacrificial agent.
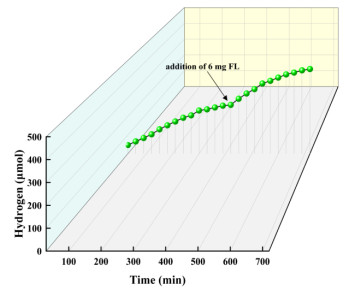
The stability of the catalytic system is critical. In Fig. 10, the hydrogen evolution rate of the reaction system is decreased with irradiation time maybe due to the decomposition of the catalyst or photosensitizer[67-69]. To investigate the specific reasons for the inactivation of the system, 6 mg of photosensitizer was added to the photocatalytic system after 6 hours of irradiation. The results show that the hydrogen-production performance was significantly improved than before. Therefore, the loss of hydrogen evolution activity in the reaction system is mainly attributed to the decomposition and consumption of the photosensitizer.
The optimized results show that complexes bind to FL mainly by the hydrogen bond OH between one oxygen atom on the carbonyl group of complexes and one hydrogen atom on the benzene ring of FL. Take complex 3 and FL as an example, the optimized binding geometry in the S0 is shown in Fig. 11. The calculated lengths of OH bond between complexes 1~4 and FL are 0.2501, 0.1686, 0.2428 and 0.2433 nm in the S0, and 0.2442, 0.1645, 0.1657 and 0.2237 nm in the S1, respectively. Corresponding gaps are 0.0059, 0.0041, 0.0771 and 0.0196 nm, respectively. Such a result shows that the lengths of OH bond in S1 shorten significantly relative to S0. It is advantageous for ET from FL to complexes by the OH bond. The result also shows that the OH bond length between complex 3 and FL is shortened greatly when electrons are excited. This implies that the excited electrons can easily transfer from FL to complex 3 by the hydrogen bond, and thus 3 may obtain the most electrons from FL. Based on the idea, complex 3 should have strong hydrogen-evolution abilities relative to complexes 1, 2 and 4. Instead, the lengths of OH bond between complexes 1, 2 and FL shorten a little when electrons are excited, showing that the excited electrons transfer from FL to complexes 1 and 2 difficultly by the hydrogen bonds. Therefore, 1 and 2 should release hydrogen difficultly, in accordance with the above experimental result.
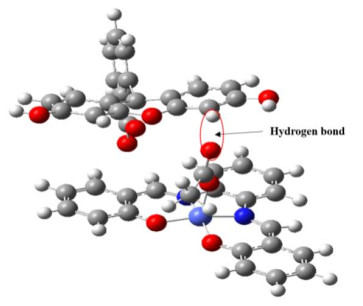
Table 1 gives the computed λ, HDA and ket. We can see that complex 3 shows the greatest ket. This indicates that the speed of ET between complex 3 and FL is much faster than those between the other complexes and FL. Such a process may lead to a great number of electrons staying on 3 by the light irradiation. Large quantities of H+ in the aqueous solution around complex 3 can then capture the electrons, easily causing H2 released. We predict that the H2 amount by 3 should be the most. Meanwhile, we predict that the order of H2 amount is 3 > 4 > 2 > 1, in accordance with our experimental result.
 DownLoad:
CSV
DownLoad:
CSV
| λ1/kJ·mol–1 | λ2/kJ·mol–1 | λtot/kJ·mol–1 | HAD/kJ·mol–1 | ket/s-1 | |
| 1 | 52.338 | 0.061 | 26.200 | 35.267 | 1.46×1018 |
| 2 | 62.427 | 0.273 | 31.350 | 35.477 | 3.26×1021 |
| 3 | 56.122 | 2.524 | 29.323 | 20.866 | 1.86×1032 |
| 4 | 65.374 | 1.054 | 33.214 | 42.383 | 4.212×1021 |
Complexes 1~4 with FL can be excited by the light irradiation. To explore the change of charges on the complexes, the natural charge populations of complexes in S0 and S1 were computed and shown in Table 2. The net charges on complexes 1~4 in the binding geometries are 0.0059 |e|, 0.0687 |e|, 0.0009 |e| and 0.0176 |e| in the S0, and 0.0071 |e|, 0.0792 |e|, 0.0795 |e| and 0.0297 |e| in the S1, respectively. Their gaps (ε) are 0.0012 |e|, 0.0105 |e|, 0.0786 |e| and 0.0121 |e|, respectively. This shows that complex 3 gained more electrons from FL than other complexes under light irradiation. Hence, large quantities of H+ around complex 3 can capture electrons easily, resulting in more H2 released relative to complexes 1, 2 and 4. Based on the charge viewpoint, we predict that the order of H2 amount should be 3 > 4 > 2 > 1, in accordance with the above experimental result.
DownLoad:
CSV
| Comp. | S0 | S1 | ε |
| 1 | 0.0059 | 0.0071 | 0.0012 |
| 2 | 0.0687 | 0.0792 | 0.0105 |
| 3 | 0.0009 | 0.0795 | 0.0786 |
| 4 | 0.0176 | 0.0297 | 0.0121 |
Based on the described experimental and theoretical results above, the mechanism for light-driven H2 production with complex 3 is proposed in Scheme 1. By the visible light, the excited FL created photogenerated electrons, which can transfer from FL to complex 3 rapidly by the hydrogen bond between them. The hydrogen-bond shortening in the excited state helps to ET. Meanwhile, the rapid ET is also favorable for a great number of electrons to stay on complex 3. Hence, large quantities of H+ in the solution around 3 can capture the electrons easily, resulting in more H2 released. This may be the primary reason for complex 3 with better hydrogenevolution properties. Therefore, the ET velocity and the number of ET are the key factors to the evolution of H2.
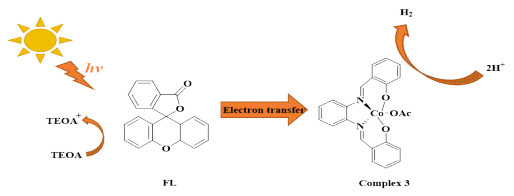
As photocatalytic hydrogen-evolution catalysts, cobalt(Ⅲ) complexes 1~4 with Schiff base were synthesized and a hydrogen-evolution system composed of Co(Ⅲ) complexes in aqueous solution, together with FL and TEOA as photosensitizer and sacrificial donor was formed. For complex 3, a maximum H2 amount of 152.3 μmol was recorded after 3 h of visible light irradiation under optimal conditions. Using DFT theory, the reason of releasing hydrogen was explained by the calculated ET rate constants, the number of ET and the change of lengths for hydrogen bonds. Besides, the mechanism for light-driven H2 production was explored by experimental and theoretical methods. This work provides a theoretical method to discover excellent catalysts, saving a lot of time and energy.
Kim, D.; Sakimoto, K. K.; Hong, D.; Yang, P. Artificial photosynthesis for sustainable fuel and chemical production. Angew. Chem. Int. Ed. 2015, 54, 3259−3266. doi: 10.1002/anie.201409116
Tachibana, Y.; Vayssieres, L.; Durrant, J. R. Artificial photosynthesis for solar water-splitting. Nat. Photonics 2012, 6, 511−518. doi: 10.1038/nphoton.2012.175
Artero, V.; Chavarot-Kerlidou, M.; Fontecave, M. Splitting water with cobalt. Angew. Chem. Int. Ed. 2011, 50, 7238−7266. doi: 10.1002/anie.201007987
Fukuzumi, S.; Lee, Y. M.; Nam, W. Thermal and photocatalytic production of hydrogen with earth-abundant metal complexes. Coord. Chem. Rev. 2018, 355, 54−73. doi: 10.1016/j.ccr.2017.07.014
Gao, C.; Wang, J.; Xu, H.; Xiong, Y. Coordination chemistry in the design of heterogeneous photocatalysts. Chem. Soc. Rev. 2017, 46, 2799−2823. doi: 10.1039/C6CS00727A
Du, P.; Eisenberg, R. Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: recent progress and future challenges. Energ. Environ. Sci. 2012, 5, 6012−6021. doi: 10.1039/c2ee03250c
Eckenhoff, W. T. Molecular catalysts of Co, Ni, Fe, and Mo for hydrogen generation in artificial photosynthetic systems. Coord. Chem. Rev. 2018, 373, 295−316. doi: 10.1016/j.ccr.2017.11.002
Eckenhoff, W. T.; McNamara, W. R.; Du, P.; Eisenberg, R. Cobalt complexes as artificial hydrogenases for the reductive side of water splitting. Biochim. Biophys. Acta 2013, 1827, 958−973. doi: 10.1016/j.bbabio.2013.05.003
Huo, J.; Zhang, Y. B.; Zou, W. Y.; Hu, X.; Deng, Q.; Chen, D. Mini-review on an engineering approach towards the selection of transition metal complex-based catalysts for photocatalytic H2 production. Catal. Sci. Technol. 2019, 9, 2716−2727. doi: 10.1039/C8CY02581A
Dong, X. Y.; Zhang, M.; Pei, R. B.; Wang, Q.; Wei, D. H.; Zang, S. Q.; Fan, Y. T.; Mak, T. C. A crystalline copper(II) coordination polymer for the efficient visible-light-driven generation of hydrogen. Angew. Chem. Int. Ed. 2016, 55, 2113−2117. doi: 10.1002/anie.201510380
Zhong, M.; Li, H.; Chen, J.; Tao, L.; Li, C.; Yang, Q. Cooperative activation of cobalt-salen complexes for epoxide hydration promoted on flexible porous organic frameworks. Chem. Eur. J. 2017, 23, 11504−11508. doi: 10.1002/chem.201702810
Song, T.; Zhang, L.; Zhang, P.; Zeng, J.; Wang, T.; Ali, A.; Zeng, H. Stable and improved visible-light photocatalytic hydrogen evolution using copper(II)-organic frameworks: engineering the crystal structures. J. Mater. Chem. A 2017, 5, 6013−6018. doi: 10.1039/C7TA00095B
Pullen, S.; Fei, H.; Orthaber, A.; Cohen, S. M.; Ott, S. Enhanced photochemical hydrogen production by a molecular diiron catalyst incorporated into a metal-organic framework. J. Am. Chem. Soc. 2013, 135, 16997−17003. doi: 10.1021/ja407176p
Hooe, S. L.; Rheingold, A. L.; Machan, C. W. Electrocatalytic reduction of dioxygen to hydrogen peroxide by a molecular manganese complex with a bipyridine-containing Schiff base ligand. J. Am. Chem. Soc. 2018, 140, 3232−3241. doi: 10.1021/jacs.7b09027
Han, Z.; McNamara, W. R.; Eum, M. S.; Holland, P. L.; Eisenberg, R. A nickel thiolate catalyst for the long-lived photocatalytic production of hydrogen in a noble-metal-free system. Angew. Chem. Int. Ed. 2012, 51, 1667−1770. doi: 10.1002/anie.201107329
Zhang, D. Modification of the optical and electronic properties of TiO2 by n anion-doping for augmentation of the visible light assisted photocatalytic performance. Chin. J. Struct. Chem. 2018, 59, 1353–1361. doi: 10.1134/S0022476618060148
Li, C. B.; Gong, P.; Yang, Y.; Wang, H. Y. Cobalt(II)-salen complexes for photocatalytic hydrogen production in noble metal-free molecular systems. Catal. Lett. 2018, 148, 3158−3164. doi: 10.1007/s10562-018-2509-y
Hogue, D. R. W.; Schott, O.; Hanan, G. S.; Brooker, S. A smorgasbord of 17 cobalt complexes active for photocatalytic hydrogen evolution. Chem. Eur. J. 2018, 24, 9820−9832. doi: 10.1002/chem.201800396
Asraf, M. A.; Younus, H. A.; Ezugwu, C. I.; Mehta, A.; Verpoort, F. Cobalt salophen complexes for light-driven water oxidation. Catal. Sci. Technol. 2016, 6, 4271−4282. doi: 10.1039/C5CY02157J
Huang, Y.; Zhang, B. Active cocatalysts for photocatalytic hydrogen evolution derived from nickel or cobalt amine complexes. Angew. Chem. Int. Ed. 2017, 56, 14804−14806. doi: 10.1002/anie.201708844
Ishizuka, T.; Watanabe, A.; Kotani, H.; Hong, D.; Satonaka, K.; Wada, T.; Shiota, Y.; Yoshizawa, K.; Ohara, K.; Yamaguchi, K.; Kato, S.; Fukuzumi, S.; Kojima, T. Homogeneous photocatalytic water oxidation with a dinuclear Co(III)-pyridylmethylamine complex. Inorg. Chem. 2016, 55, 1154−1164. doi: 10.1021/acs.inorgchem.5b02336
Banerjee, A.; Frontera, A.; Chattopadhyay, S. Methylene spacer regulated variation in molecular and crystalline architectures of cobalt(Ⅲ) complexes with reduced Schiff base ligands: a combined experimental and theoretical study. Dalton Trans. 2019, 48, 11433−11447. doi: 10.1039/C9DT01818B
Zhao, X.; Wang, P.; Liang, G.; Smith, N.; Hill, K.; Donnadieu, B.; Webster, C. E. Enhanced catalytic hydrogen evolution in neutral water by a cobalt complex with softer polypyridyl ligand. Angew. Chem. Int. Ed. 2020, 59, 12694−12697. doi: 10.1002/anie.202002640
Sasaki, Y.; Kato, H.; Kudo, A. [Co(bpy)3] (3+/2+) and [Co(phen)3] (3+/2+) electron mediators for overall water splitting under sunlight irradiation using Z-scheme photocatalyst system. J. Am. Chem. Soc. 2013, 135, 5441−5449. doi: 10.1021/ja400238r
Khandelwal, S.; Zamader, A.; Nagayach, V.; Dolui, D.; Mir, A. Q.; Dutta, A. Inclusion of peripheral basic groups activates dormant cobalt-based molecular complexes for catalytic H2 evolution in water. ACS Catal. 2019, 9, 2334−2344. doi: 10.1021/acscatal.8b04640
Elgrishi, N.; Kurtz, D. A.; Dempsey, J. L. Reaction parameters influencing cobalt hydride formation kinetics: implications for benchmarking H2-evolution catalysts. J. Am. Chem. Soc. 2017, 139, 239−244. doi: 10.1021/jacs.6b10148
Cheng, D.; Negreiros, F. R.; Apra, E.; Fortunelli, A. Computational approaches to the chemical conversion of carbon dioxide. ChemSusChem. 2013, 6, 944−965. doi: 10.1002/cssc.201200872
Zhang, L.; Lin, C. Y.; Zhang, D.; Gong, L.; Zhu, Y.; Zhao, Z.; Xu, Q.; Li, H.; Xia, Z. Guiding principles for designing highly efficient metal-free carbon catalysts. Adv. Mater. 2019, 31, e1805252. doi: 10.1002/adma.201805252
Rhodes, B.; Rowling, S.; Tidswell, P.; Woodward, S.; Brown, S. M. Aerobic epoxidation via alkyl-2-oxocyclopentanecarboxylate co-oxidation with cobalt or manganese Jacobsen-type catalysts. J. Mol. Catal. A: Chem. 1997, 116, 375−384. doi: 10.1016/S1381-1169(96)00360-3
Felicio, R. C.; Cavalheiro, E. T. G.; Dockal, E. R. Preparation, characterization and thermogravimetric studies of [N, N΄-cis-1, 2-cyclohexylene bis(salicylideneaminato)] cobalt(II) and [N, N′-(±)-trans-1, 2-cyclo-hexylene bis(salicylideneaminato)] cobalt(II). Polyhedron 2001, 20, 261−268. doi: 10.1016/S0277-5387(00)00620-3
Abdulghani, A. J.; Khaleel, A. M. Preparation and characterization of di-, tri-, and tetranuclear Schiff base complexes derived from diamines and 3, 4-dihydroxybenzaldehyde. Bioinorg. Chem. Appl. 2013, 2013, 277−306.
Liu, W. Q.; Zhou, S. L.; Fan, M. Z. Synthesis and crystal structure of a dinuclear Cu(Ⅱ) complex based on a carboxyl-substituted 1H-1, 2, 3-triazole and its DNA cleavage activity. Chin. J. Struct. Chem. 2015, 34, 917–924.
Li, H.; Xi, D.; Niu, Y.; Wang, C.; Xu, F.; Liang, L.; Xu. P. Design, synthesis and biological evaluation of cobalt(II)-Schiff base complexes as ATP-noncompetitive MEK1 inhibitors. J. Inorg. Biochem. 2019, 195, 174−181. doi: 10.1016/j.jinorgbio.2019.03.022
Dong, J.; Wang, M.; Zhang, P.; Yang, S.; Liu, J.; Li, X.; Sun, L. Promoting effect of electrostatic interaction between a cobalt catalyst and a xanthene dye on visible-light-driven electron transfer and hydrogen production. J. Phys. Chem. C 2011, 115, 15089−15096. doi: 10.1021/jp2040778
Dzygiel, P.; Reeve, T. B.; Piarulli, U.; Krupicka, M.; Tvaroska, I.; Gennari, C. Resolution of racemic N-benzyl-amino acids by liquid-liquid extraction: a practical method using a lipophilic chiral cobalt(III) salen complex and mechanistic studies. Eur. J. Org. Chem. 2008, 7, 1253−1264.
Kennedy, B. J.; Fallon, G. D.; Gatehouse, B. M. K. C.; Murray, K. S. Spin-state differences and spin crossover in five-coordinate lewis base adducts of cobalt(I1) Schiff base complexes. Structure of the high-spin (N, N΄-o -phenylenebis(salicylaldiminato)) cobalt(II)-2-methylimidazole. Inorg. Chem. 1984, 23, 580−588. doi: 10.1021/ic00173a019
Wöltinger, J.; Bäckvall, J. E.; Zsigmond, Â. Zeolite-encapsulated cobalt salophen complexes as efficient oxygen-activating catalysts in palladium-catalyzed aerobic 1, 4-oxidation of 1, 3-dienes. Chem. Eur. J. 1999, 5, 1460−1467. doi: 10.1002/(SICI)1521-3765(19990503)5:5<1460::AID-CHEM1460>3.0.CO;2-I
Chen, H.; Sun, Z.; Ye, S.; Lu, D.; Du, P. Molecular cobalt-salen complexes as novel cocatalysts for highly efficient photo-catalytic hydrogen production over a CdS nanorod photosensitizer under visible light. J. Mater. Chem. A 2015, 3, 15729−15737. doi: 10.1039/C5TA03515E
Jain, S.; Venkatasubbaiah, K.; Jones, C. W.; Davis, R. J. Factors influencing recyclability of Co(III)-salen catalysts in the hydrolytic kinetic resolution of epichlorohydrin. J. Mol. Catal. A Chem. 2010, 316, 8−15. doi: 10.1016/j.molcata.2009.10.025
Fan, G. Z.; Zhao, H. T.; Duan, Z. X.; Fang, T.; Wan, M. H.; He, L. N. A novel method to synthesize diphenyl carbonate from carbon dioxide and phenol in the presence of methanol. Catal. Sci. Technol. 2011, 1, 1138−1141. doi: 10.1039/c1cy00208b
Fast, A.; Esfandiari, N. M.; Blum, S. A. Small number of active sites and single-locus kinetics revealed in (salph) Co-catalyzed ethylene oxide polymerization. ACS Catal. 2013, 3, 2150−2153. doi: 10.1021/cs400640g
Peretti, K. L.; Ajiro, H.; Cohen, C. T.; Lobkovsky, E. B.; Coates, G. W. A highly active, isospecific cobalt catalyst for propylene oxide polymerization. J. Am. Chem. Soc. 2005, 127, 11566−11567. doi: 10.1021/ja053451y
Shyu, S. G.; Tseng, C. K.; Chang, C. K.; Chen, H. P.; Liu, H. C.; Twu, J. Effect of metal salen complex in the base catalyzed catalytic reaction between carbon dioxide and epoxides. J. Chin. Chem. Soc. 2012, 59, 443−451. doi: 10.1002/jccs.201100670
Joseph, T.; Sawant, D. P.; Gopinath, C. S.; Halligudi, S. B. Zeolite encapsulated ruthenium and cobalt Schiff base complexes catalyzed allylic oxidation of α-pinene. J. Mol. Catal. A: Chem. 2002, 184, 289−299. doi: 10.1016/S1381-1169(02)00010-9
Jain, S.; Venkatasubbaiah, K.; Jones, C. W.; Davis, R. J. Factors influencing recyclability of Co(III)-salen catalysts in the hydrolytic kinetic resolution of epichlorohydrin. J. Mol. Catal. A: Chem. 2010, 316, 8−15. doi: 10.1016/j.molcata.2009.10.025
Chen, H.; Sun, Z.; Ye, S.; Lu, D.; Du, P. Molecular cobalt-salen complexes as novel cocatalysts for highly efficient photocatalytic hydrogen production over a CdS nanorod photosensitizer under visible light. J. Mat. Chem. A 2015, 3, 15729−15737. doi: 10.1039/C5TA03515E
Kumar, D. N.; Garg, B. S. Some new cobalt(II) complexes: synthesis, characterization and thermal studies. J. Therm. Anal. Calorim. 2002, 69, 607−616. doi: 10.1023/A:1019976226610
Sakamoto, R.; Masaaki, O.; Fukita, N.; Takahashi, K. Copper(II) compounds extended by 5-carboxysalicylaldehyde and its Schiff bases: interplay of two metal-binding sites and intermolecular stacking contributing to their network and bulk structures. Bull. Chem. Soc. Jpn. 1998, 71, 2365−4701. doi: 10.1246/bcsj.71.2365
Percy, G. C.; Thornton, D. A. Infrared spectra of N-aryl salicylaldimine complexes substituted in both aryl rings. J. Inorg. Nucl. Chem. 1973, 35, 2319−2327. doi: 10.1016/0022-1902(73)80296-9
Ueno, K.; Martell, A. E. Infrared studies on synthetic oxygen carriers. J. Phys. Chem. 1956, 60, 1270−1275. doi: 10.1021/j150543a029
Felicio, R. C.; Cavalheiro, E. T. G.; Dockal, E. R. Preparation, characterization and thermogravimetric studies of [N, N΄-cis-1, 2-cyclohexylene bis(salicylideneaminato)]cobalt(II) and [N, N΄-(±)-trans-1, 2-cyclo-hexylene bis(salicylideneaminato)]cobalt(II). Polyhedron 2011, 20, 261−268.
Miao, T.; Liao, S.; Qian, L.; Zheng, K.; Ji, L. Electronic structures, DNA-binding and spectral properties of Co(III) complexes [Co(bpy)2(L)]3+ (L = pip, odhip, hnoip). Biophys. Chem. 2009, 140, 1−8. doi: 10.1016/j.bpc.2008.11.007
Vincenzo, B.; Maurizio, C. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995−2001. doi: 10.1021/jp9716997
Maurizio, C.; Nadia, R.; Giovanni, S.; Vincenzo, B. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669−681. doi: 10.1002/jcc.10189
Kuntz, I. D. Structure-based strategies for drug design and discovery. Science 1992, 257, 1078−1082. doi: 10.1126/science.257.5073.1078
Brian, K. S.; Dale, L. B.; Irwin, D. K. Molecular docking using shape descriptors. J. Comp. Chem. 1992, 13, 380−397. doi: 10.1002/jcc.540130311
Mats, H. M.; Ulf, R.; Geometry, reduction potential, and reorganization energy of the binuclear CuA site, studied by density functional theory. J. Am. Chem. Soc. 2001, 123, 7866−7876. doi: 10.1021/ja010315u
Prabha, S.; Marcus, R. A. Electron-transfer reactions in proteins: electronic coupling in myoglobin. J. Chem. Phys. B 1993, 97, 6111−6114. doi: 10.1021/j100125a004
Robert, J. C.; Marshall, D. N. Generalization of the Mulliken-Hush treatment for the calculation of electron transfer matrix elements. Chem. Phys. Lett. 1996, 249, 15−19. doi: 10.1016/0009-2614(95)01310-5
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A. Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 16, Revision C. 01, Gaussian, Inc. : Wallingford CT 2019.
Dong, J.; Wang, M.; Zhang, P.; Yang, S.; Liu, J.; Li, X.; Sun, L. Promoting effect of electrostatic interaction between a cobalt catalyst and a xanthene dye on visible-light-driven electron transfer and hydrogen production. J. Phys. Chem. C 2011, 115, 15089−15096. doi: 10.1021/jp2040778
Chen, H.; Sun, Z.; Ye, S.; Lu, D.; Du, P. Molecular cobalt-salen complexes as novel cocatalysts for highly efficient photocatalytic gydrogen production over a CdS nanorod photosensitizer under visible light. J. Mater. Chem. A 2015, 3, 15729−15737. doi: 10.1039/C5TA03515E
Fu, X.; Wang, J.; Huang, D.; Meng, S.; Zhang, Z.; Li, L.; Miao, T.; Chen, S. Trace amount of SnO2-decorated ZnSn(OH)6 as highly efficient photocatalyst for decomposition of gaseous benzene: synthesis, photocatalytic activity, and the unrevealed synergistic effect between ZnSn(OH)6 and SnO2. ACS Catal. 2015, 6, 957−968.
Lin, C.; Wang, H.; Liu, S.; Li, C.; Chu, B.; Yan, Q. Preparation of magnetic Co0.5Zn0.5Fe2O4/AgBr hybrids for the visible-light driven degradation of methyl orange. Mater. Sci. Semicond. Process 2017, 73, 67−71.
Wang, P.; Liang, G.; Reddy, M. R.; Long, M.; Driskill, K.; Lyons, C.; Donnadieu, B.; Bollinger, J. C.; Webster, C. E.; Zhao, X. Electronic and steric tuning of catalytic H2 evolution by cobalt complexes with pentadentate polypyridyl-amine ligands. J. Am. Chem. Soc. 2018, 140, 9219−9229. doi: 10.1021/jacs.8b05108
Irfan, R. M.; Jiang, D.; Sun, Z.; Lu, D.; Du, P. Enhanced photocatalytic H2 production on CdS nanorods with simple molecular bidentate cobalt complexes as cocatalysts under visible light. Dalton Trans. 2016, 45, 12897−12905. doi: 10.1039/C6DT02148D
Xu, J. X.; Yuan, Y.; Zou, S.; Chen, O.; Zhang, D. A divide-and conquer strategy for quantification of light absorption, scattering, and emission properties of fluorescent nanomaterials in solutions. Anal. Chem. 2019, 91, 8540−8548. doi: 10.1021/acs.analchem.9b01803
Natali, M. Elucidating the key role of pH on light-driven hydrogen evolution by a molecular cobalt catalyst. ACS Catal. 2017, 7, 1330−1339. doi: 10.1021/acscatal.6b03087
Cao, S. W.; Liu, X. F.; Yuan, Y. P.; Zhang, Z. Y.; Fang, J.; Loo, S. C. J.; Barber, J.; Sum, T. C.; Xue, C. Artificial photosynthetic hydrogen evolution over g-C3N4 nanosheets coupled with cobaloxime. Phys. Chem. Chem. Phys. 2013, 15, 18363−18366. doi: 10.1039/c3cp53350f

Figure 3 Absorption spectra of complexes 1~4 and FL at room temperature in MeCN: H2O = 1:1

Figure 5 Comparison of photocatalytic performance of H2 evolution for complexes 1~4. Reaction conditions: 25 mg photocatalyst, 12 mg FL, 90 mL water, and 10 mL TEOA; 300 W xenon lamp (λ ≥ 400 nm); pH = 7

Figure 6 Effect of FL content on hydrogen generation. Reaction conditions: 25 mg complex 3, different contents of PS, 90 mL water, and 10 mL TEOA; 300 W xenon lamp (λ ≥ 400 nm); pH = 7

Figure 7 Effect of the pH value of the medium on hydrogen generation. Reaction conditions: 25 mg complex 3, 12 mg FL, 90 mL water, and 10 mL TEOA; 300 W xenon lamp (λ ≥ 400 nm)

Figure 8 CVs of (a) 1.0 mM complexes and (b) the existence of 1.0 mM FL measured in 0.1 M Bu4NPF6 DMF solution. The scanning rate was 100 mV/s. Electrode setup: glassy carbon working electrode, Pt wire counter electrode, Ag/AgCl reference electrode

Figure 9 Effect of electron sacrificial agent on hydrogen evolution. Reaction conditions: 25 mg complex 3, 12 mg FL, 90 mL water, and additions of 10 mL TEOA or nothing; 300 W xenon lamp (λ ≥ 400 nm); pH = 7

Figure 10 Time courses of photocatalytic H2 evolution with a supplement of complex 3 and FL during reaction. Reaction conditions: 25 mg complex 3, 12 mg FL, 90 mL water, and 10 mL TEOA; 300 W xenon lamp (λ ≥ 400 nm); pH = 7

Figure 11 Optimized binding geometry of complex 3 with FL in the S0 and the main hydrogen bond OH
Table 1. Calculated λ1 (Complex), λ2 (FL), λtot, HDA and ket for Complexes 1~4
| λ1/kJ·mol–1 | λ2/kJ·mol–1 | λtot/kJ·mol–1 | HAD/kJ·mol–1 | ket/s-1 | |
| 1 | 52.338 | 0.061 | 26.200 | 35.267 | 1.46×1018 |
| 2 | 62.427 | 0.273 | 31.350 | 35.477 | 3.26×1021 |
| 3 | 56.122 | 2.524 | 29.323 | 20.866 | 1.86×1032 |
| 4 | 65.374 | 1.054 | 33.214 | 42.383 | 4.212×1021 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Natural Charge Populations (in |e|) of Complexes 1~4 in the Binding Geometries in the S0 and S1 and Gaps (ε)
| Comp. | S0 | S1 | ε |
| 1 | 0.0059 | 0.0071 | 0.0012 |
| 2 | 0.0687 | 0.0792 | 0.0105 |
| 3 | 0.0009 | 0.0795 | 0.0786 |
| 4 | 0.0176 | 0.0297 | 0.0121 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



