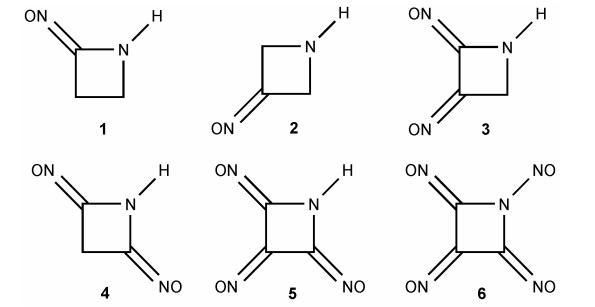
Figure 1.
Structures of azetidine derivatives designed in this paper

The search for new high energy density compounds (HEDCs) is ongoing, which have been used widely for both military and civilian applications[1-8]. In the last decades, strained polynitro cyclic compounds have attracted a lot of attentions for their high energy density and low vulnerability. RDX (hexahydro-1,3,5-trinitro-1,3,5-trizine) and HMX (1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane) belong to this class[9-13]. Such materials may find application in advanced propellants and explosive formulations. The derivatives of azetidines with strained four-member heterocyclic ring have made foray in the area of melt castable explosive since 1990s, particularly 1,3,3-trinitroazetidine (TNAZ)[14]. It is particularly attractive because of some excellent characters including low sensitiveity, easy management, and steam castability. Its melting point is 101 ℃ with density=1.84 g/cm3, and it is thermally stable (240.10 ℃) and solid at room temperature[15]. However, TNZA is with a somewhat lower output in energy than RDX and HMX[16]. Considering the importance of detonation performance (detonation velocity and detonation pressure), the way to improve them of azetidine derivatives is urgent for development.
In general, high-energy materials have large numbers of nitrogen atoms, so called "high nitrogen" compounds. This character can derive energy from the dissociation reactions of energetic N−N and C−N bonds[17, 18]. High-energy-density compounds with polynitro groups are an important class of energetic materials[19-23]. The presence of nitro groups tends to decrease the heats of formation but contributes markedly to energetic performance. Herein is why we are interested in azetidine derivatives, which contain high percentage of both oxygen and nitrogen atoms, and low amounts of carbon and hydrogen atoms. The high nitrogen content can lead to high crystal density, accompanied with increased denotation performance. The high oxygen content has an advantage in combustion reactions. For these reasons, another nitrogen-containing functional group interested in for this study is nitroso groups (=NO). The main difference among nitroso group and nitro group is the nitrogen content. In addition, the C=N bond is stronger than the C–N bond and can introduce more stability.
Therefore, we replaced the hydrogen atoms of azetidine molecule with nitroso groups to design new potential HEDCs. It is expected that our results provide some useful information for synthesis of polynitroso derivatives of azetidine.
All calculations were performed at B3PW91/6-311+G(d,p)//MP2/6-311+G(d,p) level by using Gaussian 03 program packages[24-26], along with the standard Gaussian basis set 6-311+G(d,p). The method can deal with the system with large dynamic electron correlation effectively and provide more accurate electronic energy, which also emerged in title compounds, especially for the polynitrososubstituted azetidine[27]. Harmonic vibrational analyses were performed at the same level of theory to confirm the structural nature. Fig. 1 listed all object molecules.
The heats of formation (HOFs) are needed in the calculation of detonation energy. In present paper, the atomization reaction[28] was applied to calculate the HOFs of title compounds. For 1, 2, 3, 4, and 5, reaction (1) was used. For 6 compound, reaction (2) was used. HOFs are obtained via equation (3)~(5).
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
The pyrolysis mechanism and thermal stability can be evaluated using the bond dissociation energy (BDE), the difference between the energies of the parent molecule and the corresponding radicals in the unimolecular bond dissociation[30-32]. The gasphase BDE for a bond
|
|
(6) |
|
|
(7) |
The bond dissociation energy with zero-point energy (ZPE) correction can be calculated by Eq. (8):
|
|
(8) |
Where ΔZPE is the difference between the ZPEs of products and the reactants.
For each title compound, explosive reaction is designed in terms of the maximal exothermal principle, that is, all the N atoms were turned into N2, the O atoms react with H atoms to give H2O at first, and then form CO2 with C atom. If the number of O atoms is more than needed to oxidize H and C atoms, redundant O atoms will be converted into O2. If the number of atoms is not enough to satisfy full oxidation of H and C atoms, and the remaining of H atoms will convert into H2O, and the C atoms will exist as solid-state C. For the CHNO explosives, the detonation velocity (D) and detonation pressure (P) were estimated by Kamlet-Jacobs equation (9) and (10)[33].
|
|
(9) |
|
|
(10) |
N is moles of detonation gases per gram explosive,
It is well-known that evaluation for detonation performance of energetic materials requires the knowledge of HOFs. Moreover, HOFs are of great importance for researchers involved in thermal chemistry. Higher are the molecular HOFs, more energy stores the molecule. However, to obtain HOFs via experimental methods is extremely hazardous and difficult, so, theoretical studies is particularly important and quite necessary. In our work, an atomization reaction was applied to calculate the HOFs of title compounds. Table 1 presented the calculated total energies, the zeropoint energies, the values of thermal correction, and HOFs of azetidine derivatives at the B3PW91/6-311+G(d,p)//MP2/6-311+G(d,p) level.
 DownLoad:
CSV
DownLoad:
CSV
| Compounds | E0 | ZPE | ∆HT | HOF |
| 1 | –301.71590 | 0.08252 | 0.08931 | 488.67 |
| 2 | –301.73099 | 0.08301 | 0.08982 | 448.93 |
| 3 | –430.45254 | 0.06863 | 0.07711 | 483.00 |
| 4 | –430.45443 | 0.06901 | 0.07712 | 481.23 |
| 5 | –559.13438 | 0.05495 | 0.06476 | 620.76 |
| 6 | –688.40261 | 0.05115 | 0.06297 | 2296.64 |
Inspecting the values of HOFs, we found that all azetidine derivatives have high positive HOFs, which become larger as the numbers of nitroso group increased. Obviously, the contributions of nitroso group on HOFs of azetidine derivatives meet the group additivity rule. The higher the molecular HOFs, the more the molecule stores energy. Furthermore, for isomers, such as 1 and 2, it is found that the HOF value of 1 is higher than 2 about 40kJ/mol, which indicated that the thermochemical stability of 2 is better than 1. The reason is the steric interaction is weaker in 2 than that in 1, which increases the molecular stability compared to 1. For 3 and 4, the HOF value of 3 is higher than 4 about 2kJ/mol, which is caused by the steric hindrance of nitroso groups. The nitroso groups in 3 are closer each other than that in 4 insulting in the steric hindrance effect which raised HOF value of 3, meanwhile, the molecule stability is dropped down. Although high HOFs is negative for thermal stability, it can improve detonation energy. In addition, we also should notice that HOFs value in this paper are the superior limit in practice for their gas-phases state, as pointed out by Politzer and cooperators[34].
The sensitivity and stability of energetic compounds are directly relevant to the bond strength, which is commonly described by BDE[32, 35, 36]. In general, stronger are the weakest bond, more stable is the energetic material. Bond order reflect the electron accumulations in bonding region, and they can provide us with detailed information about the chemical bond. That is to say that the less bond order a bond has, the easier the bond breaks. In previous papers, the method had been applied successfully[37-39]. So the weakest bond order is selected as the trigger bond on the basis of the results of populations analysis[40] in this paper. The bond order and bond dissociation energies of trigger bond at B3PW91/6-311+G(d,p) level are listed in Table 2.
DownLoad:
CSV
| Compound | Bond Order | BDE | BDEZPE |
| 1 | 1.00 | 165.65 | 151.58 |
| 2 | 0.98 | 195.87 | 180.23 |
| 3 | 1.10 | 215.73 | 196.05 |
| 4 | 1.32 | 227.44 | 210.56 |
| 5 | 1.57 | 313.44 | 295.00 |
| 6 | 1.21 | 261.03 | 238.04 |
It can be found from Table 2 that the BDE values without zero point energy correction are larger than BDEZPE (including zero point energies correction). However, the sequence of dissociation energies are not affected by the zero-point energies. It should be pointed out that BDEs of the trigger bonds in all these azetidine derivatives are lower than that of TATB (276.93kJ/mol). However, their BDEZPE values are over 120kJ/mol, which means these compounds sufficient stability request of explosives. The BDEZPE of 2 is larger than 1, which indicate 2 is more stable than 1 kinetically consistent with the results from HOFs calculations in last section. For 3 and 4, the BDEZPE of 4 is larger than 3 about 10 kJ/mol. This is because that all atoms except hydrogen atoms of 4 are in the same plane, as well as strong conjugate action exists in 4 system. The correlation between the bond orders and the bond dissociation energies are positive from 1 to 5. However, 6 has lower bond order but higher BDEZPE value compared to others. This shows that the kinetic stability of azetidine derivatives is not determined simply by the bond orders, and the BDEZPE must be considered. Furthermore, on the consideration of the unobservable in experiment of bond order, the prediction based on bond dissociation energies is more reliable.
Detonation velocity and detonation pressure are two important performance parameters for high energetic compounds. Several empirical methods have been applied to estimate these parameters[41], although the errors of density lead to D and P somewhat deviating from experiments[42]. The Kamlet-Jacobs equation has been proved to be reliable[43] and used in this paper. The molecular density, heats of detonation, detonation velocity and detonation pressure were listed in Table 3 with that of RDX and HMX for comparison.
DownLoad:
CSV
| Compound | ρ | Q | D | P |
| 1 | 1.79 | 1196.11 | 6.95 | 21.39 |
| 2 | 1.79 | 1169.55 | 6.92 | 21.22 |
| 3 | 2.07 | 1217.76 | 8.33 | 33.42 |
| 4 | 2.08 | 1216.87 | 8.35 | 33.64 |
| 5 | 2.29 | 1288.68 | 9.36 | 44.42 |
| 6 | 2.39 | 1874.54 | 10.80 | 60.70 |
| RDX | 1.78(1.82) | 1591.03 | 8.87(8.75) | 34.67(34.00) |
| HMX | 1.88(1.91) | 1633.90 | 9.28(9.10) | 39.19(39.00) |
As is evident in Table 3, the ρ of azetidine derivatives rises as introduction of substituent groups. The maximal and least ρ value are 1.93 and 1.47 g/cm3, respectively. Inspecting the ρ values of 1 and 2, it is found that the ρ value of 1 is close to that of 2. As the introduction of nitroso groups, the molecular density are rising from 1.79 to 2.39, which is better than that of HMX. As we known, a molecular density better than 2.0g/cm3 is desirable until now in the field of explosive, therefore, the molecule 6 is glamorous from the viewpoint of density. For D and P, we find that the detonation performances of 5 and 6 are better than RDX, especially 6 compound, the D and P have over HMX, one of the most widely used energetic compound in the field of high-performance explosive. Although 3 and 4 also have good detonation performance, they can not meet the requirement as HEDCs (D = 9.0 km/s and P = 40 GPa). In addition, 1 and 2 have the worst D and P in all compounds, and further research is not reasonable. 1,3,3-Trinitroazetidine (TNAZ) is a well characterized high energy density compound, and the explosive heat, the molecular density, the detonation velocity and the detonation pressure are 1160J/g, 1.84g/cm3, 9.60km/s, and 36.4GPa, respectively[44]. Compared to TNAZ, the tri-substituted derivative 5 and tetra-substituted derivative 6 have better detonation characters and others are inferior. Evidently, NO group is more effective to improve the molecular density than nitro group, because dinitroso-substituted derivatives have larger density than trinitro-substituted derivatives. On the consideration the importance of molecular density for explosive characters, nitroso group is a better energetic group than nitro group. In summary, 5 and 6 are recommended as candidates of HEDCs in this study.
Based on our calculations, it is shown that all azetidine derivatives possess large positive HOFs, which increase with the introduction of nitroso groups. The predicted detonation velocities and detonation pressures indicate that nitroso group is an effective substituent group to enhancing detonation performance. Particularly 5 (D = 9.36km/s and P = 44.42GPa) and 6 (D = 10.80km/s and P = 60.70GPa) may be the promising candidates of HEDCs.
The kinetic stability and pyrolysis mechanism were evaluated by using bond dissociation energies. For azetidine derivatives, the homolysis of C−N is the initial step in explosion reactions. Moreover, the BDEs of all molecules are over 120kJ/mol, which meet the criterion of HEDCs. The steric hindrance has also influence on the molecular stability.
Huynh M. H. V.; Hiskey M. A.; Chavez D. E.; Naud D. L.; Gilardi R. D. Synthesis, characterization, and energetic properties of diazido heteroaromatic high-nitrogen C−N Compound. J. Am. Chem. Soc. 2005, 127, 12537−12543. doi: 10.1021/ja0509735
Gutowski K. E.; Rogers R. D.; Dixon D. A. Accurate Thermochemical properties for energetic materials applications. Ⅱ. Heats of formation of imidazolium-, 1,2,4-Triazolium-, and tetrazolium-based energetic salts from isodesmic and lattice energy calculations. J. Phys. Chem. B 2007, 111, 4788−4800. doi: 10.1021/jp066420d
Li B.; Zhou M.; Peng J.; Li L.; Guo Y. Theoretical calculations about nitro-substituted pyridine as high-energy-density compounds (HEDCs). J. Mol. Model. 2019, 25, 23−28. doi: 10.1007/s00894-018-3904-4
Shu X.; Tian Y.; Song G.; Zhang H.; Kang B.; Zhang C.; Liu Y.; Liu X.; Sun J. Thermal expansion and theoretical density of 2, 2′, 4, 4′, 6, 6′-hexanitrostilbene. J. Mater. Sci. 2011, 46, 2536−2540. doi: 10.1007/s10853-010-5105-0
Li Y.; Feng X.; Liu H.; Hao J.; Redfern S. A. T.; Lei W.; Liu D.; Ma Y. Route to high-energy density polymeric nitrogen t-N via He-N compounds. Nat. Commun. 2018, 9, 722−728. doi: 10.1038/s41467-018-03200-4
Wu J.; Huang Y.; Yang L.; Geng D.; Wang F.; Wang H.; Chen L. Reactive molecular dynamics simulations of the thermal decomposition mechanism of 1,3,3-trinitroazetidine (TNAZ). ChemPhysChem 2018, 19, 2683−2695. doi: 10.1002/cphc.201800550
Liu F. L.; Liu Y.; Zhang L.; Wu Y. M. A dodecahedrane-like molecule C12H12B8 with uncommon Th symmetry. Chinese J. Struct. Chem. 2012, 31, 677−682.
Mei Z.; Li X. H.; Cui H. L.; Wang H. X.; Zhang R. Z. Theoretical studies on the structure and detonation properties of a furazan- based energetic macrocycle compound. Chinese J. Struct. Chem. 2016, 35, 16−24.
Smith G. D.; Bharadwaj R. K. Quantum chemistry based force field for simulations of HMX. J. Phys. Chem. B 1999, 103, 3570−3575. doi: 10.1021/jp984599p
Brill T. B.; Gongwer P. E.; Williams G. K. Thermal decomposition of energetic materials. 66. kinetic compensation effects in HMX, RDX, and NTO. J. Phys. Chem. 1994, 98, 12242−12247. doi: 10.1021/j100098a020
Alavi G.; Chung M.; Lichwa J.; D'Alessio M.; Ray C. The fate and transport of RDX, HMX, TNT and DNT in the volcanic soils of Hawaii: A laboratory and modeling study. J. Hazard. Mater. 2011, 185, 1600−1604. doi: 10.1016/j.jhazmat.2010.10.039
Ariyarathna T.; Ballentine M.; Vlahos P.; Smith R. W.; Cooper C.; Bohlke J. K.; Fallis S.; Groshens T. J.; Tobias C. Tracing the cycling and fate of the munition, Hexahydro-1,3,5-trinitro-1,3,5-triazine in a simulated sandy coastal marine habitat with a stable isotopic tracer, (15)N-[RDX]. Sci. Total. Environ. 2019, 647, 369−378. doi: 10.1016/j.scitotenv.2018.07.404
Eberly J. O.; Mayo M. L.; Carr M. R.; Crocker F. H.; Indest K. J. Detection of hexahydro-1,3-5-trinitro-1,3,5-triazine (RDX) with a microbial sensor. J. Gen. Appl. Microbiol. 2019, 64, 139−144.
Archibald T. G.; Gilardi R.; Baum K.; George C. Synthesis and x-ray crystal structure of 1,3,3-trinitroazetidine. J. Org. Chem. 1990, 55, 2920−2924. doi: 10.1021/jo00296a066
Thompson C. A.; Rice J. K.; Russell T. P.; Seminario J. M.; Politzer P. Vibrational analysis of 1,3,3-trinitroazetidine using matrix isolation infrared spectroscopy and quantum chemical calculations. J. Phys. Chem. A 1997, 101, 7742−7748. doi: 10.1021/jp971173m
Sikder N.; Sikder A. K.; Bulakh N. R.; Gandhe B. R. 1,3,3-Trinitroazetidine (TNAZ), a melt-cast explosive: synthesis, characterization and thermal behaviour. J. Hazard. Mater. 2004, 113, 35−43. doi: 10.1016/j.jhazmat.2004.06.002
Hammerl A.; Klapötke T. M.; Nöth H.; Warchhold M.; Holl G.; Kaiser M.; Ticmanis U. [N2H5]+2[N4C−NN−CN4]2-: A new high-nitrogen high-energetic material. Inorg. Chem. 2001, 40, 3570−3575. doi: 10.1021/ic010063y
Chavez D. E.; Hiskey M. A. 1,2,4,5-tetrazine based energetic materials. J. Energetic Mater. 1999, 17, 357−377. doi: 10.1080/07370659908201796
De Vries L.; Winstein S. Neighboring carbon and hydrogen. XXXIX. 1 Complex rearrangements of bridged ions. Rearrangement leading to the bird-cage hydrocarbon1. J. Am. Chem. Soc. 1960, 82, 5363−5376. doi: 10.1021/ja01505a023
Liebman J. F.; Greenberg A. A survey of strained organic molecules. Chem. Rev. 1976, 76, 311−365. doi: 10.1021/cr60301a002
Marchand A. P.; Wu A. Syntheses of new substituted pentacyclo[5.4. 0.02, 6.03, 10.05, 9]undecanes: a novel synthesis of hexacyclo[6.2. 1.13, 6.02, 7.04, 10.05, 9]dodecane (1,3-bishomopentaprismane). J. Org. Chem. 1986, 51, 1897−1900. doi: 10.1021/jo00360a046
Nielsen A. T.; Nissan R. A.; Vanderah D. J.; Coon C. L.; Gilardi R. D.; George C. F.; Flippen-Anderson J. Polyazapolycyclics by condensation of aldehydes with amines. 2. Formation of 2,4,6,8,10,12-hexabenzyl-2,4,6,8,10,12-hexaazatetracyclo [5.5. 0.05. 9.03, 11] dodecanes from glyoxal and benzylamines. J. Org. Chem. 1990, 55, 1459−1466. doi: 10.1021/jo00292a015
Schulman J. M.; Disch R. L. Ab initio heats of formation of medium-sized hydrocarbons. The heat of formation of dodecahedrane. J. Am. Chem. Soc. 1984, 106, 1202−1204. doi: 10.1021/ja00317a005
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian, Inc., Pittsburgh PA 2003, Gaussian 03, Revision B. 01.
Hehre W. J.; Ditchfield R.; Pople J. A. Self−Consistent molecular orbital methods. XII. Further extensions of Gaussian−Type basis sets for use in molecular orbital studies of organic olecules. J. Chem. Phys. 1972, 56, 2257−2261. doi: 10.1063/1.1677527
Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785−789. doi: 10.1103/PhysRevB.37.785
Schütz M.; Hetzer G.; Werner H.-J. Low-order scaling local electron correlation methods. I. Linear scaling local MP2. J. Chem. Phys. 1999, 111, 5691−5705. doi: 10.1063/1.479957
Curtiss L. A.; Raghavachari K.; Redfern P. C.; Stefanov B. B. Assessment of complete basis set methods for calculation of enthalpies of formation. J. Chem. Phys. 1998, 108, 692−697. doi: 10.1063/1.475442
Curtiss L. A.; Raghavachari K.; Redfern P. C.; Pople J. A. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J. Chem. Phys. 1997, 106, 1063−1079. doi: 10.1063/1.473182
Shao J.; Cheng X.; Yang X. Density functional calculations of bond dissociation energies for removal of the nitrogen dioxide moiety in some nitroaromatic molecules. J. Mol. Struct. THEOCHEM 2005, 755, 127−130. doi: 10.1016/j.theochem.2005.08.008
Politzer P.; Lane P. Comparison of density functional calculations of C–NO2, N–NO2 and C–NF2 dissociation energies. J. Mol. Struct. THEOCHEM 1996, 388, 51−55.
Harris N. J.; Lammertsma K. Ab initio density functional computations of conformations and bond dissociation energies for hexahydro-1,3,5-trinitro-1,3,5-triazine. J. Am. Chem. Soc. 1997, 119, 6583−6589. doi: 10.1021/ja970392i
Kamlet M. J.; Jacobs S. J. Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives. J. Chem. Phys. 1968, 48, 23−35. doi: 10.1063/1.1667908
Politzer P.; Ma Y.; Lane P.; Concha M. C. Computational prediction of standard gas, liquid, and solid-phase heats of formation and heats of vaporization and sublimation. Int. J. Quantum Chem. 2005, 105, 341−347. doi: 10.1002/qua.20709
Owens F. J. Calculation of energy barriers for bond rupture in some energetic molecules. J. Mol. Struct. THEOCHEM 1996, 370, 11−16. doi: 10.1016/S0166-1280(96)04673-8
Guo L. Density functional study of structural and electronic properties of GaPn (2 ≤ n ≤ 12) clusters. J. Mater. Sci. 2010, 45, 3381−3387. doi: 10.1007/s10853-010-4361-3
Fan X.-W.; Ju X.-H. Theoretical studies on four-membered ring compounds with NF2, ONO2, N3, and NO2 groups. J. Comput. Chem. 2008, 29, 505−513. doi: 10.1002/jcc.20809
Rice B. M.; Sahu S.; Owens F. J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. J. Mol. Struct. THEOCHEM 2002, 583, 69−72. doi: 10.1016/S0166-1280(01)00782-5
Zhang J.; Xiao H. Computational studies on the infrared vibrational spectra, thermodynamic properties, detonation properties, and pyrolysis mechanism of octanitrocubane. J. Chem. Phys. 2002, 116, 10674−10683. doi: 10.1063/1.1479136
Mulliken R. S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833−1840. doi: 10.1063/1.1740588
Keshavarz M. H.; Pouretedal H. R. Simple empirical method for prediction of impact sensitivity of selected class of explosives. J. Hazard. Mater. 2005, 124, 27−33. doi: 10.1016/j.jhazmat.2005.05.009
Bulat F.; Toro-Labbé A.; Brinck T.; Murray J.; Politzer P. Quantitative analysis of molecular surfaces: areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679−1691. doi: 10.1007/s00894-010-0692-x
Gálvez-Ruiz J. C.; Holl G.; Karaghiosoff K.; Klapötke T. M.; Löhnwitz K.; Mayer P.; Nöth H.; Polborn K.; Rohbogner C. J.; Suter M.; Weigand J. J. Derivatives of 1,5-Diamino-1H-tetrazole: A new family of energetic heterocyclic-based salts. Inorg. Chem. 2005, 44, 4237−4253. doi: 10.1021/ic050104g
Axenrod T.; Watnick C.; Yazdekhasti H.; Dave P. R. Synthesis of 1,3,3-trinitroazetidine. Tetrahedron Lett. 1993, 34, 677−6680. doi: 10.1016/S0040-4039(00)61650-7
Table 1. Total Energy (E0, a.u.), Zero-point Energy (ZPE, a.u.), Heats of Formation (HOF, kJ/mol) and Thermal Correction (∆HT, a.u.) Calculated at B3PW91/6-311+G(d,p)//MP2/6-311+G(d,p) Level
| Compounds | E0 | ZPE | ∆HT | HOF |
| 1 | –301.71590 | 0.08252 | 0.08931 | 488.67 |
| 2 | –301.73099 | 0.08301 | 0.08982 | 448.93 |
| 3 | –430.45254 | 0.06863 | 0.07711 | 483.00 |
| 4 | –430.45443 | 0.06901 | 0.07712 | 481.23 |
| 5 | –559.13438 | 0.05495 | 0.06476 | 620.76 |
| 6 | –688.40261 | 0.05115 | 0.06297 | 2296.64 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Bond Dissociation Energies (kJ/mol) and Bond Orders of N−NO2 Calculated at the B3PW91/6-311+G(d,p)//MP2/6-311+G(d,p) Level
| Compound | Bond Order | BDE | BDEZPE |
| 1 | 1.00 | 165.65 | 151.58 |
| 2 | 0.98 | 195.87 | 180.23 |
| 3 | 1.10 | 215.73 | 196.05 |
| 4 | 1.32 | 227.44 | 210.56 |
| 5 | 1.57 | 313.44 | 295.00 |
| 6 | 1.21 | 261.03 | 238.04 |
下载: 导出CSV
Table 3. Molecular Density (g/cm3), Explosive Heats (kJ/g), Detonation Pressure (GPa) and Detonation Velocity (km/s) of Azetidine Derivatives Together with RDX and HMX
| Compound | ρ | Q | D | P |
| 1 | 1.79 | 1196.11 | 6.95 | 21.39 |
| 2 | 1.79 | 1169.55 | 6.92 | 21.22 |
| 3 | 2.07 | 1217.76 | 8.33 | 33.42 |
| 4 | 2.08 | 1216.87 | 8.35 | 33.64 |
| 5 | 2.29 | 1288.68 | 9.36 | 44.42 |
| 6 | 2.39 | 1874.54 | 10.80 | 60.70 |
| RDX | 1.78(1.82) | 1591.03 | 8.87(8.75) | 34.67(34.00) |
| HMX | 1.88(1.91) | 1633.90 | 9.28(9.10) | 39.19(39.00) |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
