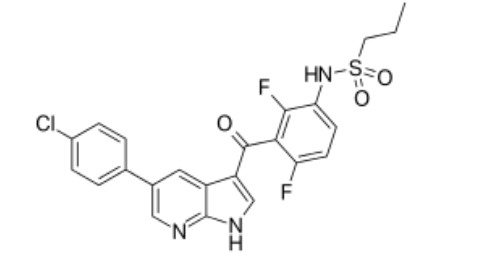
Figure 1.
Chemical structure of Vemurafenib
Theoretical Study of Adsorption Behavior of Vemurafenib Drug over BNNT(5,5-9) as a Factor of Drug Delivery: a DFT Study
Sheikhi MASOOME , Shahab SIYAMAK , Alhosseini Almodarresiyeh HORA , Khaleghian MEHRNOOSH , Kumar RAKESH , Strogova ALEKSANDRA

To control the side effects and toxicity of drugs is an essential and challenging task and it tends to be overcome by sending chemical drugs to the target region of the body. Target drug delivery at the site of action can be done by methods like direct injection, catheter, gene gun, etc. But these systems show direct delivery, invasiveness is not patient convenient and expensive to carry out in many cases[1-5]. Presently towards this direction efforts are made to develop TDDS (target drug delivery systems) which include chemical, physical and biological modifications or functionalization with or without the use of carriers. Functionalization of drug delivery agents with carbon nanotubes (CNTs) has received a lot of consideration in research because of their exceptional physical properties. Functionalization of drug delivery agents is one of the safe and established methods enabling the agents to interact better with the drugs[6-9].
One targeted cancer drug Vemurafenib (Zelboraf) which is also known as cancer growth blocker (Fig. 1) is used to treat a type of skin cancer called melanoma that has spread to other parts of the body or cannot be removed by surgery, and that has a certain type of abnormal "BRAF" gene[10]. A suitable understanding of the interaction behavior of drug molecules with BNNTs is a fundamental step for the development of nanoscale drug delivery vehicles. During the last decade, many attempts have been made to analyze the interactions of BNNTs with different biomolecules and drugs by using DFT calculations. Nanotubes are the almost concerning nanovectors among nanomaterials which have attracted attention for drug delivery. Boron nitride nanotubes (BNNTs) have pulled considerable interest in nanomedicine due to their brilliant characteristics[11]. These nanotubes are useful emerging materials for biomedical, therapeutic, and diagnostic applications because of their good biocompatibility and no cytotoxicity[12]. Larger band gap of BNNTs, compared to carbon nanotubes, makes these compounds suitable for working in hazardous and high temperature environments of biological systems[13-17]. BNNTs are also chemically inert and resistant to oxidation and corrosion. Charge distribution in the BN nanotubes is asymmetric in B–N bonds and due to higher electronegativity of nitrogen the electron density of B is attracted towards N atoms. Therefore, the B–N bonds have a partially ionic character that creates a large band gap around 5.5 eV between the valence and conduction bands. Structural parameters, energies and electronic properties of the complex can be easily calculated after geometry optimization[18, 19]. Mukhopadhyay and co-workers have been studied the sensitivity of boron nitride nanotube (BNNT) toward biomolecules including amino acids namely tryptophan (Trp), aspartic acid (Asp) and arginine (Arg) by DFT calculations[20]. In this study we have carried out the investigation of interaction between BNNTs and Vemurafenib based on DFT method. We have investigated frontier molecular orbitals, quantum-chemical molecular descriptors, MEP analysis, natural charge, chemical shift tensors and charge transfer analysis according to NBO analysis, electronic structure and excited states.
DFT studies can provide more useful information on the interaction between nanotubes and delivered drug molecu-les[19-21]. Recently, we have studied the adsorption anticancer drug Syndros[22], Resveratrol[23] and Alectinib[24] over CNTs.
In the present work, the adsorption of Vemurafenib on the sidewall SWCNT has been performed based on the DFT method.
In this research, the non-bonded interaction between BNNT(5,5-9) with an anticancer drug Vemurafenib in the solvent water was investigated. The Polarized Continuum Model (PCM)[25] was used for the calculations of solvent effect. The quantum chemical calculations have been performed using density functional theory (DFT) calculations at the M062X/6-31G* level of theory by the Gaussian 09W software[26] for optimization of the molecule Vemurafenib, BNNT(5,5-9) and complex BNNT(5,5-9)/Vemurafenib. The adsorption energy (Ead)[21] was computed using the following equation:
|
|
where EBNNT(5,5-9)/Vemurafenib, EBNNT(5,5-9) and EVemurafenib are energies of BNNT(5,5-9) with the adsorbed Vemurafenib, BNNT(5,5-9) and the compound Vemurafenib, respectively.
The molecular orbital (MO) calculations of the investigated molecular systems such as EHOMO, ELUMO, energy gap between LUMO and HOMO (Eg = ELUMO –EHOMO) were also carried out. The optimized structures, HOMO, LUMO and MEP surfaces were visualized using the GaussView 05 software[27].
In addition, the adsorption effects of the molecule Vemurafenib over BNNT(5,5-9) on the natural charge and the chemical shielding tensors[22, 23] such as chemical shift isotropic (σiso) and chemical shift anisotropic (σaniso) were computed at the M062X/6-31G* level of theory. The σiso and σaniso parameters were calculated using the following equations, respectively:
|
|
(1) |
|
|
(2) |
The three parameters such as σ11, σ22, σ33 show chemical shielding interaction in three dimensions. The calculations of electronic transitions of the molecule Vemurafenib and the complex BNNT(5,5-9)/Vemurafenib were performed by TD-DFT method[23]. The electronic structure of the title com-pounds was also studied by using NBO analysis[28] at the M062X/6-31G* level in order to understand the hyperconju-gative interactions and charge delocalization.
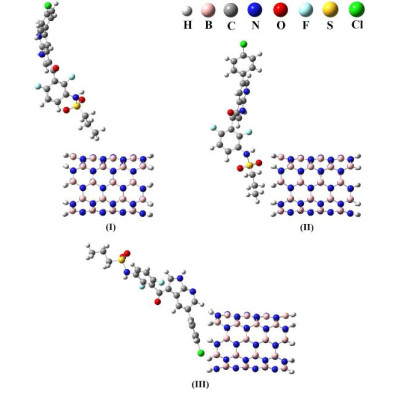
In the first step of calculations, interaction between the Vemurafenib drug with the BNNT(5,5-9) nanotube has been carried out. We chose three configurations for investigation that Vemurafenib is located in different situations around the BNNT(5,5-9) nanotube. All the three configurations (states I, II and III) were optimized at the PM6 method[22] (Fig. 2) and their energy values (HF) are reported in Table 1. According to obtained results, the lowest energy value was observed for state II.
 DownLoad:
CSV
DownLoad:
CSV
| Systems | HF (Hartree) |
| State I | –3.6435114 |
| State II | –3.6471549 |
| State III | –3.6463678 |
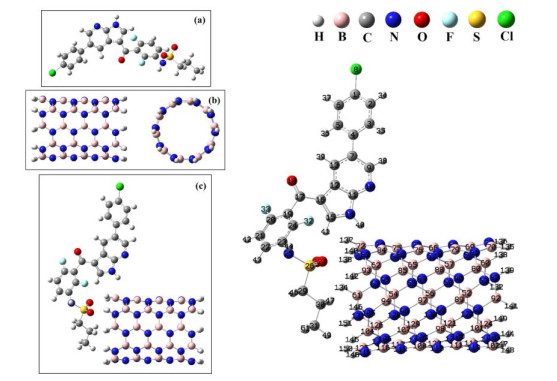
In the next step, we have optimized the compounds Vemurafenib, BNNT(5,5-9) and complex BNNT(5,5-9)/Vemurafenib (state II) using M062X/6-31G* level of theory in a solvent water. The optimized structures of the title compounds are shown in Fig. 3.
The thermochemical parameters were calculated for the molecules Vemurafenib, BNNT(5,5-9) and complex BNNT(5,5-9)/Vemurafenib with M062X/6-31G* level of theory that are shown in Table 2. According to the summari-zed results in Table 2, with the adsorption of the molecule Vemurafenib on BNNT(5,5-9), the Thermal, Gibbs and Enthalpy energy values decrease. Energy values reflect the reduced reactivity and increase the stability of Vemurafenib drug in non-bonded reaction with BNNT(5,5-9).
DownLoad:
CSV
| Parameter | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib |
| E+G (Hartree) | –2334.2965 | –3199.1035 | 5534.6514 |
| E+H (Hartree) | –2334.2018 | –3198.9859 | 5534.3940 |
| E+T (Hartree) | –2334.2028 | –3198.9869 | 5534.5237 |
| E+ZPE (Hartree) | –2334.2318 | –3199.0351 | 5534.1382 |
| S (cal/mol·K) | 199.189 | 247.353 | 300.498 |
Geometrical parameters play a significant role in interpreting the attachment of drug to nanotubes in drug delivery systems. The calculated bond lengths of optimized Vemurafenib, BNNT(5,5-9), and BNNT(5,5-9)/Vemurafenib complex at the binding sites are reported in Table 3. According to observed results, some geometrical parameters of Vemurafenib and BNNT are changed after interaction between them. The lengths of B(72)–N(73), B(72)–N(74), N(83)–B(84), B(72)–H(137), N(73)–H(138), and N(83)–H(140) in the open end of BNNT(5,5-9) are 1.420, 1.453, 1.445, 1.195, 1.012, 1.012 Å, and change to 1.428, 1.450, 1.442, 1.192, 1.015, 1.016 Å, respectively after the adsorption of Vemurafenib drug on BNNT(5,5-9) (see Table 3). These bonds show the most changes in BNNT(5,5-9) nanotube during interaction with Vemurafenib drug. The optimized structure of Vemurafenib drug has the C(19)–C(24), C(24)–F(32), N(25)–S(26), S(26)–O(28), S(26)–C(29), N(14)–H(40), C(15)–H(41), C(30)–H(48) bond lengths of 1.391, 1.337, 1.696, 1.459, 1.873, 1.011, 1.081, 1.094 Å, while with adsorption over BNNT(5,5-9) nanotube they change to be 1.387, 1.339, 1.687, 1.463, 1.782, 1.014, 1.083, 1.096 Å, respectively. According to these results, the geometry of structures Vemurafenib and BNNT(5,5-9) change after the interaction of the two compounds and the formation of the complex, although these changes are not basic.
DownLoad:
CSV
| Bond length | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib |
| N(14)–C(15) | 1.355 | - | 1.354 |
| C(19)–C(24) | 1.391 | - | 1.387 |
| C(23)–C(24) | 1.391 | - | 1.392 |
| C(23)–N(25) | 1.426 | - | 1.427 |
| C(24)–F(32) | 1.337 | - | 1.339 |
| N(25)–S(26) | 1.696 | - | 1.687 |
| S(26)–O(27) | 1.462 | - | 1.462 |
| S(26)–O(28) | 1.459 | - | 1.463 |
| S(26)–C(29) | 1.873 | - | 1.782 |
| C(29)–C(30) | 1.527 | - | 1.527 |
| C(30)–C(31) | 1.526 | - | 1.526 |
| N(14)–H(40) | 1.011 | - | 1.014 |
| C(15)–H(41) | 1.081 | - | 1.083 |
| N(25)–H(44) | 1.021 | - | 1.020 |
| C(30)–H(47) | 1.094 | - | 1.095 |
| C(30)–H(48) | 1.094 | - | 1.096 |
| C(31)–H(49) | 1.092 | - | 1.091 |
| B(61)–N(63) | - | 1.453 | 1.452 |
| B(72)–N(73) | - | 1.420 | 1.428 |
| B(72)–N(74) | - | 1.453 | 1.450 |
| N(83)–B(84) | - | 1.445 | 1.442 |
| N(83)–B(93) | - | 1.420 | 1.419 |
| B(93)–N(95) | - | 1.453 | 1.454 |
| B(61)–N(131) | - | 1.420 | 1.420 |
| B(94)–N(105) | - | 1.445 | 1.445 |
| B(104)–N(105) | - | 1.420 | 1.419 |
| B(125)–N(115) | - | 1.420 | 1.420 |
| B(125)–N(127) | - | 1.453 | 1.453 |
| B(126)–N(131) | - | 1.445 | 1.446 |
| B(61)–H(134) | - | 1.195 | 1.196 |
| B(72)–H(137) | - | 1.195 | 1.192 |
| N(73)–H(138) | - | 1.012 | 1.015 |
| N(83)–H(140) | - | 1.012 | 1.016 |
| B(93)–H(142) | - | 1.195 | 1.196 |
| N(105)–H(146) | - | 1.012 | 1.012 |
| N(131)–H(151) | - | 1.012 | 1.012 |
NBO analysis is used for investigating intra- and intermolecular bonding and interaction between bonds in molecules[29]. The electron delocalization from donor orbitals (full NBOs) to acceptor orbitals (empty NBOs) shows a conjugative electron transfer process between them[29]. While electron transfers from donor orbital (i) to acceptor orbital (j) and delocalization process i → j, the stabilization energy E(2) is computed[30]:
|
|
(4) |
The stabilization energy (E(2)) shows the amount of the participation of electrons in the resonance between atoms of the molecules[29]. The larger E(2) means that the electron donor has a lot of tendency for donation to electron acceptor[28]. The NBO analysis for complex BNNT(5,5-9)/Vemurafenib has been performed using M062X/6-31G* level of theory and the results are summarized in Table 4. According to obtained results, the π → π*, π → σ*, σ → σ*, σ → π*, n → σ*, π* → π*, π* → σ* and σ* → σ* transitions take place between Vemurafenib and BNNT(5,5-9). The lone pairs (n) of the O(27) and O(28) atoms in the compound Vemurafenib overlaps with the anti-bonding orbitals σ* of BNNT(5,5-9) that include n1(O(27)) → σ*(N(83)–H(140), n2(O(28)) → σ*(N(73)–H(138)) interactions with the highest stabilization energies (E(2)) of 3.18 and 3.29 kcal/mol, respectively, rather than other transitions Vemurafenib → BNNT. The other important transitions of Vemurafenib → BNNT include π(C(11)–N(14)) → π*(B(72)–N(74)), π*(C(11)–N(14)) → π*(B(72)–N(74)), π*(C(15)–C(16)) → π*(B(72)–N(74)), σ*(S(26)–O(27)) → σ*(N(83)–H(140)), σ*(S(26)–O(28)) → σ*(N(73)–H(138)) interactions with stabilization energies (E(2)) about 0.32, 0.38, 0.47, 0.76 and 0.76 kcal/mol, respectively. The maximum stabilization energy (E(2)) in charge transfer from BNNT to Vemurafenib is observed for the π(B(62)–N(73)) → σ*(N(14)–H(40)) transition with maximum resonance energy (E(2)) about 2.95 kcal/mol observed in charge transfer from BNNT to Vemurafenib. The other important transitions of BNNT → Vemurafenib include σ(B(61)–H(134)) → σ*(C(30)–H(48)), σ(B(125)–H(150)) → σ*(C(31)–H(49)) and π*(B(61)–N(63)) → σ*(C(30)–H(48)) interactions with resonance energies to be about 0.29, 0.48 and 0.30 kcal/mol, respectively. Thus, Vemurafenib and BNNT(5,5-9) act as both electron donor and acceptor; therefore, charge transfer takes place between them in BNNT(5,5)/Vemurafenib.
DownLoad:
CSV
| Donor (i) | Acceptor (j) | E(2)a (kcal/mol) | E(j)-E(i)b (a.u.) | F(i, j)c (a.u.) |
| π(C(11)–N(14)) | π*(B(72)–N(74)) | 0.32 | 0.49 | 0.011 |
| σ(N(14)–H(40)) | σ*(B(72)–N(73)) | 0.07 | 1.31 | 0.009 |
| π*(B(72)–N(74)) | 0.17 | 0.87 | 0.011 | |
| σ(C(15)–H(41)) | σ*(N(73)–H(138)) | 0.12 | 1.20 | 0.011 |
| σ(C(30)–H(48)) | π*(B(61)–N(63)) | 0.15 | 0.66 | 0.009 |
| σ(C(31)–H(49)) | σ*(N(115)–H(148)) | 0.27 | 1.12 | 0.016 |
| σ*(B(125)–N(127)) | 0.09 | 1.08 | 0.009 | |
| π*(B(125)–N(127)) | 0.17 | 0.66 | 0.010 | |
| n1(O(27)) | σ*(N(83) H(140)) | 3.18 | 1.46 | 0.061 |
| σ*(B(93)–N(95)) | 0.10 | 1.41 | 0.011 | |
| n2(O(27)) | σ*(B(72)–H(137)) | 0.17 | 0.96 | 0.012 |
| σ*(N(83)–H(140)) | 0.23 | 0.94 | 0.014 | |
| n1(O(28)) | σ*(B(72)–N(73)) | 0.05 | 1.42 | 0.008 |
| σ*(B(72)–N(74)) | 0.22 | 1.41 | 0.016 | |
| n2(O(28)) | σ*(N(73)–H(138)) | 3.29 | 1.45 | 0.062 |
| σ*(B(72)–N(74)) | 0.11 | 0.90 | 0.009 | |
| π*(C(11)–N(14)) | π*(B(72)–N(74)) | 0.38 | 0.13 | 0.009 |
| π*(C(15)–C(16)) | π*(B(72)–N(74)) | 0.47 | 0.04 | 0.008 |
| σ*(S(26)–O(27)) | σ*(N(83)–H(140)) | 0.76 | 0.24 | 0.042 |
| σ*(S(26)–O(28)) | σ*(N(73)–H(138)) | 0.76 | 0.23 | 0.040 |
| π(B(61)–N(63)) | σ*(N(14)–H(40)) | 0.09 | 0.85 | 0.008 |
| σ(B(61)–H(134)) | σ*(C(30)–H(48)) | 0.29 | 1.01 | 0.015 |
| π(B(62)–N(73)) | σ*(C(11)–N(14)) | 0.14 | 0.84 | 0.010 |
| σ*(N(14)–H(40)) | 2.95 | 0.87 | 0.047 | |
| σ*(C(15)–C(16)) | 0.09 | 0.96 | 0.009 | |
| σ*(C(15)–H(41)) | 0.09 | 0.88 | 0.008 | |
| π(B(72)–N(73)) | σ*(N(14)–H(40)) | 0.09 | 1.30 | 0.009 |
| σ(B(72)–H(137)) | π*(C(15)–C(16)) | 0.11 | 0.52 | 0.007 |
| σ*(S(26)–O(27)) | 0.18 | 0.80 | 0.011 | |
| σ*(S(26)–O(28)) | 0.15 | 0.80 | 0.010 | |
| σ*(S(26)–C(29)) | 0.11 | 0.67 | 0.008 | |
| σ(N(73)–H(138)) | σ*(C(15)–H(41)) | 0.12 | 1.24 | 0.011 |
| σ*(S(26)–O(28)) | 0.10 | 1.02 | 0.009 | |
| σ(N(83)–B(84)) | σ*(S(26)–O(27)) | 0.06 | 1.10 | 0.008 |
| σ(N(83)–H(140)) | σ*(S(26)–C(27)) | 0.09 | 1.02 | 0.009 |
| π(N(115)–B(116)) | σ*(C(31)–H(49)) | 0.16 | 1.32 | 0.013 |
| σ*(C(31)–H(49)) | 0.10 | 0.88 | 0.009 | |
| σ(N(115)–H(148)) | σ*(C(31)–H(49)) | 0.18 | 1.24 | 0.013 |
| σ(B(125)–H(150)) | σ*(C(31)–H(49)) | 0.48 | 1.03 | 0.020 |
| π*(B(61)–N(63)) | σ*(C(30)–H(48)) | 0.30 | 0.44 | 0.030 |
| π*(B(72)–N(74)) | σ*(N(25)–S(26)) | 0.15 | 0.07 | 0.006 |
| π*(B(93)–N(95)) | σ*(C(30)–H(47)) | 0.06 | 0.43 | 0.013 |
| π*(B(125)–N(127)) | σ*(C(31)–H(49)) | 0.19 | 0.46 | 0.025 |
| a E(2) Energy of hyperconjucative interactions. b Energy difference between donor and acceptor i and j NBO orbitals. c F(i, j) is the Fock matrix element between i and j NBO orbitals. | ||||
The frontier molecular orbitals (FMO) including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are important parameters in chemical reactions[31]. The HOMO and LUMO energies exhibit the capability to donate and obtain an electron, respectively. The molecular orbitals have an important role in charge transfer process in molecular systems. The energy gap (Eg) between HOMO and LUMO orbitals is a significant parameter in specifying electronic transport properties in molecules[32]. We have studied the non-bonded intermolecular interaction effects between Vemurafenib drug and BNNT(5,5-9) on the electronic properties. The calculated results are reported in Table 5.
DownLoad:
CSV
| Property | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib |
| Dipole moment (Debye) | 6.78 | 0.00 | 11.62 |
| E(Hartree) | –2334.61294 | –3199.69538 | –5534.32742 |
| Ead (Hartree) | – | – | –0.0191 |
| EHOMO (eV) | –7.66 | –7.95 | –7.53 |
| ELUMO (eV) | –1.06 | 0.94 | –0.89 |
| Eg (eV) | 6.60 | 7.01 | 6.64 |
| I (eV) | 7.66 | 7.95 | 7.53 |
| A (eV) | 1.06 | 0.94 | 0.89 |
| χ (eV) | 4.36 | 4.44 | 4.21 |
| η (eV) | 3.30 | 3.50 | 3.32 |
| μ (eV) | –4.63 | –4.44 | –4.21 |
| ω (eV) | 3.24 | 2.81 | 2.66 |
| S (eV) | 0.15 | 0.14 | 0.15 |
The adsorption energy (Ead) of the Vemurafenib drug over BNNT(5,5-9) has a negative amount of about –0.0191 eV; therefore, the reaction is exothermic (Table 5).
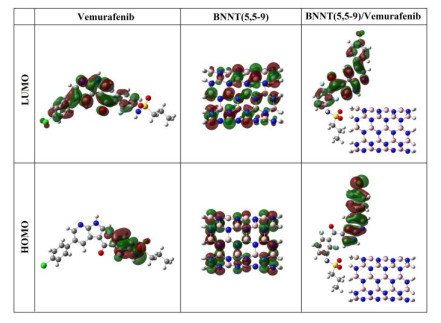
As shown in Fig. 4, the electron density in HOMO orbital of the compound Vemurafenib is mainly situated on the double bonds (-C=C-) of fluorine-substituted benzene ring, nitrogen and oxygen atom (N–H) of amine group, fluorine atoms and oxygen atoms of SO2 group, whereas the LUMO orbital is situated on double bonds (-C=C-) of chlorine and fluorine-substituted benzene rings, oxygen atom of carbonyl group, double bonds (-C=C-) and nitrogen atoms of pyridine and pyrrole rings. Thus, most of the charge transfer from HOMO to LUMO orbital in the Vemurafenib drug is due to the contribution of π bonds and lone pairs. The HOMO and LUMO orbitals of BNNT(5,5-9) are localized on nitrogen and boron atoms, respectively. According to the obtained results, HOMO and LUMO orbitals of complex BNNT(5,5-9)/Vemurafenib mainly focus on Vemurafenib drug.
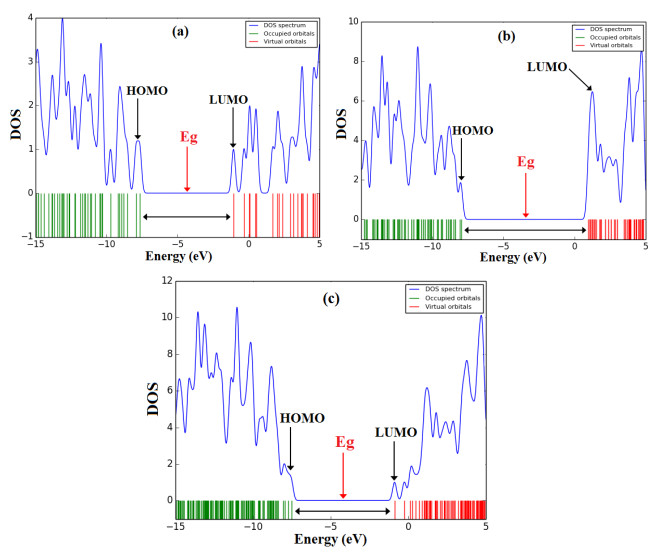
The energy gap (Eg) in BNNT(5,5-9) is 7.01 eV, whereas after the adsorption process of Vemurafenib drug on BNNT(5,5-9) (complex) decreases to 6.64 eV. This result shows a significant increase in electrical conductivity of system complex compared with the isolated BNNT(5,5-9). DOS plots[33] in Fig. 5 also shows the energy gaps of the title systems.
The quantum molecular descriptors for the compounds Vemurafenib, BNNT(5,5-9) and complex BNNT(5,5-9)/Vemurafenib consist of ionization potential (I), electron affinity (A), global hardness (η), electronegativity (χ), electro-nic chemical potential (µ), electrophilicity (ω) and chemical softness (S) are calculated according to the following equations[31]:
|
|
and they are reported in Table 5. The electronic properties of Vemurafenib change with the adsorption over BNNT (Table 5). The stability of the molecules is related to hardness which is an important parameter for detection of chemical reactivity[32]. The hardness and electronic chemical potential parameters of the complex BNNT/drug will be decreased, while electro-philicity and softness will be increased rather than isolated Vemurafenib drug. Therefore, the complex has a high chemical activity, low chemical stability and it is a soft system. Thus, it is found that the adsorption of Vemurafenib drug on BNNT(5,5-9) in the solvent water changes electronic properties.
The values of dipole moment of BNNT(5,5-9), Vemura-fenib and complex BNNT(5,5-9)/Vemurafenib are 0.00, 6.78 and 11.62 Debye, respectively (Table 7). After adsorption process, the values of dipole moment of Vemurafenib and BNNT increase from 0.00, 6.78 to 11.62 Debye, indicating a charge transfer between Vemurafenib drug and BNNT(5,5-9). The atomic charges have an important role in physical properties such as molecular polarizability, dipole moment, electronic structure and related properties of molecular systems[33]. The NBO charges for equilibrium geometry of BNNT(5,5-9), Vemurafenib and complex were calculated using the M062X/6-31G* level of theory. The calculated natural charges for selected atoms in these three molecular systems are reported in Table 6 (Atoms are numbered according to Fig. 3). The natural charges of C(24), N(25), S(26), O(27), O(28), C(29), C(31), F(32), H(41), H(44), H(48) and H(49) atoms at the compound Vemurafenib are 0.464, –0.916, 2.384, –0.994, –0.995, –0.719, –0.691, –0.335, 0.271, 0.452, 0.255 and 0.250 e, while change to 0.459, –0.936, 2.402, –0.999, –1.004, –0.710, –0.683, –0.330, 0.267, 0.465, 0.250 and 0.237 e, respectively after adsorption process of Vemurafenib on the BNNT(5,5-9). The natural charges of B(61), N(63), N(73), N(74), N(83), B(94), N(115) and B(125) atoms of BNNT(5,5-9) are 0.818, –1.217, –1.147, –1.217, –1.147, 1.194, –1.147 and 0.818 e, respectively. After adsorption of Vemurafenib on BNNT(5,5-9), the natural charges change to be 0.813, –1.216, –1.177, –0.219, –0.152, 1.193, –1.148 and 0.813 e, respectively. The change of atomic charges induces a dipole moment in the complex. Thus, it shows a charge transfer and non-bonded interaction between Vemurafenib drug and BNNT(5,5-9).
DownLoad:
CSV
| Atoms | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib | ||||||||
| Charge | CSI | CSA | Charge | CSI | CSA | Charge | CSI | CSA | |||
| N(14) | –0.557 | 105.44 | 116.82 | - | - | - | –0.558 | 98.26 | 126.29 | ||
| C(15) | 0.072 | 49.29 | 152.43 | - | - | - | 0.071 | 46.69 | 155.36 | ||
| C(23) | 0.038 | 66.62 | 128.36 | - | - | - | 0.040 | 65.24 | 126.80 | ||
| C(24) | 0.464 | 30.91 | 113.82 | - | - | - | 0.459 | 26.97 | 121.88 | ||
| N(25) | –0.916 | 137.17 | 143.47 | - | - | - | –0.936 | 147.43 | 155.12 | ||
| S(26) | 2.384 | 214.42 | 188.03 | - | - | - | 2.402 | 212.77 | 135.97 | ||
| O(27) | –0.994 | 118.02 | 98.47 | - | - | - | –0.999 | 120.24 | 69.14 | ||
| O(28) | –0.995 | 148.59 | 149.93 | - | - | - | –1.004 | 129.10 | 72.88 | ||
| C(29) | –0.719 | 135.95 | 68.85 | - | - | - | –0.710 | 132.74 | 68.67 | ||
| C(30) | –0.490 | 179.28 | 10.25 | - | - | - | –0.489 | 177.77 | 10.66 | ||
| C(31) | –0.691 | 182.49 | 21.56 | - | - | - | –0.683 | 180.39 | 19.84 | ||
| F(32) | –0.335 | 312.65 | 177.15 | - | - | - | –0.330 | 289.69 | 170.36 | ||
| F(33) | –0.330 | 295.90 | 146.47 | - | - | - | –0.331 | 296.90 | 167.99 | ||
| H(40) | 0.476 | 23.61 | 9.61 | - | - | - | 0.476 | 23.25 | 15.82 | ||
| H(41) | 0.271 | 23.97 | 8.28 | - | - | - | 0.267 | 23.11 | 9.95 | ||
| H(44) | 0.452 | 26.82 | 8.00 | - | - | - | 0.465 | 26.80 | 8.76 | ||
| H(47) | 0.257 | 30.36 | 6.61 | - | - | - | 0.255 | 30.58 | 7.28 | ||
| H(48) | 0.255 | 30.40 | 6.80 | - | - | - | 0.250 | 30.69 | 8.58 | ||
| H(49) | 0.250 | 30.89 | 11.43 | - | - | - | 0.237 | 30.65 | 12.24 | ||
| B(61) | - | - | - | 0.818 | 78.57 | 59.03 | 0.813 | 79.02 | 58.06 | ||
| B(62) | - | - | - | 1.193 | 83.03 | 37.51 | 1.193 | 82.63 | 37.42 | ||
| N(63) | - | - | - | –1.217 | 134.42 | 208.98 | –1.216 | 134.53 | 209.64 | ||
| B(72) | - | - | - | 0.818 | 78.62 | 59.67 | 0.818 | 75.55 | 65.25 | ||
| N(73) | - | - | - | –1.147 | 144.41 | 148.88 | –1.177 | 142.77 | 150.78 | ||
| N(74) | - | - | - | –1.217 | 134.67 | 210.10 | –0.219 | 133.42 | 208.04 | ||
| N(83) | - | - | - | –1.147 | 144.30 | 149.41 | –0.152 | 141.05 | 154.80 | ||
| B(93) | - | - | - | 0.818 | 78.51 | 58.77 | 0.818 | 78.39 | 59.17 | ||
| B(94) | - | - | - | 1.194 | 83.03 | 37.85 | 1.193 | 82.82 | 37.68 | ||
| B(104) | - | - | - | 0.818 | 78.68 | 58.80 | 0.818 | 79.03 | 60.05 | ||
| N(105) | - | - | - | –1.147 | 144.31 | 149.54 | –1.147 | 143.88 | 151.17 | ||
| N(115) | - | - | - | –1.147 | 144.45 | 148.84 | –1.148 | 144.03 | 149.86 | ||
| B(125) | - | - | - | 0.818 | 78.13 | 58.63 | 0.813 | 78.23 | 60.97 | ||
| B(126) | - | - | - | 1.193 | 82.97 | 37.63 | 1.192 | 83.43 | 36.81 | ||
| N(127) | - | - | - | –1.217 | 134.56 | 209.54 | –1.217 | 135.68 | 206.23 | ||
DownLoad:
CSV
| Excited state | Wavelength (nm) |
Excitation energy (eV) | Configurations composition (corresponding transition orbitals) |
Oscillator strength (f) | ||||
| S0 → S1 | 256 | 4.82 | H-9 → L (31%), H-9 → L+5 (14%), H-6 → L (18%) H-9 → L+1 (3%), H-9 → L+12 (2%), H-8 → L (2%), H-6 → L+5 (7%) |
0.00 | ||||
| S0 → S2 | 228 | 5.41 | H-1 → L (25%), H → L (15%), H → L+1 (14%), H → L+2 (17%) H-4 → L (2%), H-4 → L+6 (3%), H-3 → L+4 (2%), H-1 → L+1 (8%), H-1 → L+2 (2%) |
0.35 | ||||
| S0 → S3 | 224 | 5.53 | H-1 → L (25%), H → LU (24%), H → L+1 (24%), H-4 → L+2 (3%), H-1 → L+1 (3%), H-1 → L+6 (2%), H → L+2 (7%) |
0.32 | ||||
| S0 → S4 | 205 | 6.04 | H-1 → L (13%), H → L+2 (26%) H-7 → L (3%), H-5 → L (3%), H-4 → L (4%), H-4 → L+1 (4%), H-3 → L+4 (7%), H-1 → L+1 (4%), H-1 → L+2 (7%), H-1 → L+5 (6%), H → L+4 (2%) |
0.28 | ||||
| S0 → S5 | 202 | 6.11 | H-4 → L+4 (11%), H-3 → L+2 (19%), H → L+4 (35%), H-3 → L (6%), H-3 → L+1 (9%), H-3 → L+6 (5%), H-1 → L+4 (6%) |
0.01 | ||||
| S0 → S6 | 201 | 6.15 | H-5 → L+3 (19%), H-2 → L (35%), H-2 → L+1 (25%) H-2 → L+5 (9%) |
0.09 | ||||
| S0 → S7 | 198 | 6.24 | H-1 → L+1 (12%), H-1 → L+2 (24%), H-5 → L (6%), H-5 → L+1 (7%), H-4 → L (5%), H-3 → L+4 (5%), H-2 → L+3 (8%), H-1 → L (2%), H-1 → L+5 (5%), H → L+1 (4%), H → L+5 (2%), H → L+6 (5%) | 0.39 | ||||
| S0 → S8 | 195 | 6.32 | H-6 → L+1 (22%), H-6 → L+2 (19%), H-9 → L (3%), H-9 → L+1 (5%), H-9 → L+2 (5%), H-6 → L (8%), H-6 → L+5 (7%), H-2 → L+3 (2%) | 0.00 | ||||
| S0 → S9 | 194 | 6.36 | H-5 → L (16%), H-2 → L+3 (29%), H-5 → L+1 (9%), H-5 → L+5 (2%), H-1 → L (5%), H-1 → L+1 (5%), H-1 → L+2 (5%), H-1 → L+5 (7%) | 0.18 | ||||
| S0 → S10 | 176 | 7.03 | H-3 → L+4 (35%), H-1 → L+2 (10%), H → L (13%), H-4 → L (3%), H-4 → L+2 (5%), H-1 → L+1 (7%), H → L+2 (6%), H → L+5 (5%), H → L+6 (2%) | 1.14 | ||||
| S0 → S11 | 170 | 7.27 | H-1 → L+1 (20%), H-1 → L+2 (12%), H-1 → L+6 (13%), H → L+2 (10%), H-7 → L(4%), H-5 → L (3%), H-3 → L+2 (3%), H-3 → L+4 (5%), H-2 → L+3 (5%), H-1 → L (3%), H-1 → L+5 (3%), H → L+1 (2%) | 0.80 | ||||
| S0 → S12 | 167 | 7.41 | H-3 → L+4 (12%), H → L (18%), H → L+1 (15%), H → L+2 (12%), H → L+5(13%), H-4 → L (6%), H-4 → L+2 (3%), H-4 → L+5 (3%), H → L+6 (5%) |
0.17 | ||||
| S0 → S13 | 166 | 7.43 | H-5 → LUMO (28%), H-2 → L+3 (37%), H-5 → L+1 (5%), H-1 → L+2 (2%) | 0.61 | ||||
| S0 → S14 | 162 | 7.61 | H-3 → L (10%), H-3 → L+1 (13%), H-3 → L+2 (19%), H → L+4 (30%), H-4 → L+4 (3%), H-3 → L+6 (3%), H-1 → L+1 (3%), H-1 → L+4 (3%) | 0.58 | ||||
| S0 → S15 | 159 | 7.78 | H-4 → L+7 (30%), H → L+7 (43%), H-13 → L+7 (5%), H-7 → L+7 (2%), H-1 → L+7 (8%) | 0.00 | ||||
| S0 → S16 | 158 | 7.80 | H-5 → L+3 (60%), H-2 → L (20%), H-5 → L+5 (3%), H-4 → L+3 (3%), H-2 → L+1 (4%) |
0.60 | ||||
| S0 → S17 | 154 | 8.02 | H-4 → L+2 (16%), H-3 → L+4 (20%), H → L+6 (10%), H-7 → L (3%), H-7 → L+1 (6%), H-6 → L (2%), H-6 → L+2 (2%), H-6 → L+6 (2%), H-4 → L (3%), H-4 → L+1 (3%), H-1 → L+1 (7%), H-1 → L+5 (2%), H-1 → L+6 (5%) | 0.06 | ||||
| S0 → S18 | 152 | 8.13 | H-6 → L+2 (16%), H-6 → L+6 (10%), H-9 → L (2%), H-9 → L+2 (4%), H-9 → L+6 (2%), H-7 → L+1 (4%), H-6 → L (7%), H-4 → L (2%), H-4 → L+1 (3%), H-1 → L (5%), H-1 → L+1 (9%), H-1 → L+5 (8%), H → L+5 (3%) | 0.01 | ||||
| S0 → S19 | 150 | 8.21 | H-1 → L+5 (20%), H → L+6 (10%), H-6 → L (5%), H-6 → L+2 (8%), H-6 → L+6 (6%), H-4 → L+1 (4%), H-4 → L+2 (3%), H-3 → L+4 (5%), H-1 → L (6%), H-1 → L+1 (7%), H-1 → L+2 (3%), H → L+5 (2%) | 0.02 | ||||
| S0 → S20 | 147 | 8.42 | H-5 → L+5 (28%), H-5 → L+1 (9%), H-5 → L+3 (2%), H-4 → L (3%), H-4 → L+1 (6%), H-2 → L+1 (3%), H-2 → L+3 (7%), H-2 → L+5 (9%), H-1 → L+5 (2%), H → L (3%) | 0.19 | ||||
| *H: HOMO, L: LUMO | ||||||||
We have calculated the NMR parameters such as chemical shift isotropic (σiso) and chemical shift anisotropic (σaniso) for selected atoms in the molecules Vemurafenib, BNNT(5,5-9) and complex BNNT(5,5-9)/Vemurafenib using the M062X/6-31G* level of theory. The electronic density affects the electrostatic properties of atoms. The adsorption of Vemura-fenib drug on BNNT(5,5-9) changes the electronic densities of atoms and NMR parameters. The results of the chemical shift tensors (ppm) are reported in Table 6. The obtained results show that the values of σiso for the B(61), B(62), B(72), N(73), N(74), N(83), N(115), B(126), N(127) atoms of BNNT(5,5-9) are 78.57, 83.03, 78.62, 144.41, 134.67, 144.30, 144.45, 82.97, 134.56 ppm, respectively. After the adsorption of Vemurafenib drug on BNNT(5,5-9), these values were estimated to be about 79.02, 82.63, 75.55, 142.77, 133.42, 141.05, 144.03, 83.43 and 135.68 ppm, respectively. The title atoms exhibit the main changes because Vemurafenib in the complex is close to these atoms in BNNT. For the molecule Vemurafenib, the σiso values of N(14), C(15), C(24), N(25), O(28) and F(32) atoms are 105.44, 49.29, 30.91, 137.17, 148.59 and 312.65 ppm, respectively, but these values after the adsorption process of Vemurafenib over BNNT(5,5-9) change to 98.26, 46.69, 26.97, 147.43, 129.10 and 289.69 ppm, respectively.
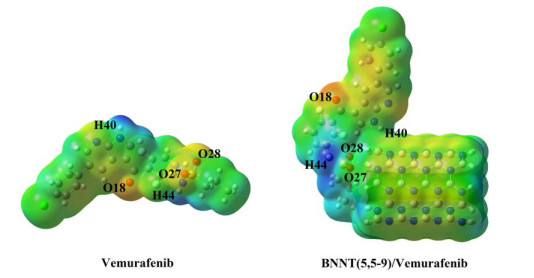
Molecular electrostatic potential (MEP) maps illustrate the electronic density in the molecules and they are used to identify sites of positive and negative electrostatic potentials surfaces through different colors[22, 23]. In MEPs, the negative sites with high electron density have red, orange or yellow colors related to electrophilic reactivity, whereas the positive regions with low electron density have blue color and they were related to nucleophilic reactivity and green color was used for neutral regions[32]. The MEPs of the molecule Vemurafenib and complex BNNT(5,5-9)/Vemurafenib were computed by DFT calculations using the M062X/6-31G* level of theory (Fig. 6) and the charge distribution was investigated. As seen from the MEP maps of Vemurafenib and complex BNNT/drug, the O(18) atom with red color has the highest electron density. The O(27) and O(28) atoms of isolated Vemurafenib have yellow color; whereas after adsorption process, the O(27) and O(28) atoms in complex BNNT/drug have green color that displays the neutral region. This color change shows interaction between drug and BNNT. The lowest electron density with blue color in the MEP map of Vemurafenib is observed for H(40) and H44 atoms; while in the complex BNNT/drug, only H(44) atom with blue color has the lowest electron density. The H(40) atom of Vemurafenib is neutral due to the interaction with BNNT.
In order to study of interaction effect of Vemurafenib with BNNT(5,5-9) on the λmax, we have calculated the UV spectra of Vemurafenib and complex BNNT(5,5-9)/Vemurafenib in the solvent water using TDM062X/6-31G* method with considering 20 excited states which are reported in Tables 7, 8 and Fig. 7. Tables 7 and 8 display the λmax, oscillator strength (f), and excitation energies (E).
DownLoad:
CSV
| Excited state | Wavelength (nm) |
Excitation energy (eV) | Configurations composition (corresponding transition orbitals) |
Oscillator strength (f) | ||||
| S0 → S1 | 256 | 4.84 | H-36 → L (12%), H-24 → L (19%), H-41 → L (3%), H-36 → L+5 (5%), H-35 → L (3%), H-33 → L (6%), H-33 → L+5 (3%), H-32 → L (4%), H-24 → L+5 (8%) | 0.00 | ||||
| S0 → S2 | 228 | 5.41 | H-1 → L (30%), H-1 → L+1 (10%), H → L (10%), H → L+1 (12%), H → L+2 (12%), H-9 → L (3%), H-4 → L+4 (2%), H-1 → L+2 (2%), H → L+3 (5%) | 0.30 | ||||
| S0 → S3 | 224 | 5.51 | H-1 → L (19%), H → L (28%), H → L+1 (26%), H-9 → L+2 (4%), H-1 → L+5 (2%), H → L+2 (8%) | 0.42 | ||||
| S0 → S4 | 205 | 6.03 | H-1 → LUMO (14%), H → L+2 (23%), H-26 → L (4%), H-9 → L (5%), H-9 → L+1 (4%), H-4 → L+4 (6%), H-1 → L+1 (6%), H-1 → L+2 (3%), H-1 → L+3 (4%), H-1 → L+5 (7%), H → L (2%), H → L+3 (3%), H → L+5 (2%) | 0.35 | ||||
| S0 → S5 | 202 | 6.11 | H-9 → L+4 (12%), H-4 → L+1 (10%), H-4 → L+2 (12%), H → L+4 (34%), H-4 → L (6%), H-4 → L+3 (6%), H-4 → L+18 (3%), H-1 → L+4 (8%) | 0.01 | ||||
| S0 → S6 | 199 | 6.22 | H-1 → L+2 (28%), H-12 → L+3 (3%), H-9 → L (3%), H-9 → L+1 (4%), H-6 → L (6%), H-6 → L+1 (5%), H-4 → L+4 (6%), H-1 → L+1 (8%), H-1 → L+5 (3%), H → L+1 (4%), H → L+5 (2%), H → L+18 (4%) | 0.37 | ||||
| S0 → S7 | 198 | 6.24 | H-12 → L+3 (14%), H-6 → L (28%), H-6 → L+1 (20%), H-12 → L+2 (4%), H-6 → L+5 (7%), H-1 → L+1 (2%), H-1 → L+2 (4%) | 0.14 | ||||
| S0 → S8 | 196 | 6.32 | H-24 → L+1 (18%), H-24 → L+2 (20%), H-33 → L+1 (2%), H-33 → L+2 (3%), H-32 → L+2 (2%), H-24 → L (7%), H-24 → L+5 (6%) | 0.02 | ||||
| S0 → S9 | 192 | 6.43 | H-12 → L (23%), H-12 → L+1 (14%), H-6 → L+3 (27%), H-13 → L (3%), H-13 → L+1 (2%), H-12 → L+5 (3%), H-6 → L+2 (6%), H-1 → L (2%), H-1 → L+5 (3%) | 0.01 | ||||
| S0 → S10 | 177 | 7.02 | H-4 → L+4 (34%), H-1 → L+2 (12%), H → L (13%), H-9 → L (4%), H-9 → L+2 (5%), H-1 → L+1 (7%), H-1 → L+5 (2%), H → L+2 (6%), H → L+5 (5%) | 1.19 | ||||
| S0 → S11 | 170 | 7.26 | H-1 → L+1 (23%), H → L+2 (13%), H-26 → L (4%), H-4 → L+4 (7%), H-1 → L (4%), H-1 → L+2 (7%), H-1 → L+3 (6%), H-1 → L+5 (3%), H-1 → L+18 (9%) | 0.59 | ||||
| S0 → S12 | 167 | 7.41 | H-4 → L+4 (12%), H → L (18%), H → L+1 (15%), H → L+5 (12%) H-9 → L (6%), H-9 → L+5 (3%), H → L+2 (5%), H → L+3 (8%), H → L+18 (3%) | 0.23 | ||||
| S0 → S13 | 164 | 7.53 | H-12 → L (26%), H-6 → L+3 (27%), H-13 → L (4%), H-12 → L+1 (3%), H-12 → L+3 (2%), H-6 → L+2 (6%), H-4 → L (2%), H-4 → L+1 (3%), H-4 → L+2 (3%), H → L+4 (5%) | 0.62 | ||||
| S0 → S14 | 162 | 7.61 | H-4 → L+1 (13%), H-4 → L+2 (11%), H → L+4 (25%), H-12 → L (5%), H-9 → L+4 (3%), H-6 → L+3 (5%), H-4 → L (9%), H-4 → L+3 (6%), H-1 → L+1 (3%), H-1 → L+4 (4%) | 0.63 | ||||
| S0 → S15 | 159 | 7.78 | H-9 → L+23 (32%), H-1 → L+23 (10%), H → L+23 (39%), H-48 → L+23 (5%), H-26 → L+23 (2%) | 0.00 | ||||
| S0 → S16 | 157 | 7.85 | H-12 → L+2 (10%), H-12 → L+3 (40%), H-6 → L (20%), H-13 → L+3 (6%), H-12 → L+5 (2%), H-6 → L+1 (5%), H-6 → L+3 (2%), H-1 → L+3 (2%) | 0.84 | ||||
| S0 → S17 | 154 | 8.01 | H-9 → L+2 (11%), H-4 → L+4 (19%), H-26 → L (3%), H-26 → L+1 (7%), H-9 → L (3%), H-9 → L+1 (2%), H-9 → L+3 (4%), H-1 → L+1 (8%), H-1 → L+5 (4%), H-1 → L+18 (4%), H → L+18 (5%) | 0.06 | ||||
| S0 → S18 | 153 | 8.10 | H-1 → L+1 (10%), H-1 → L+5 (13%), H-26 → L+1 (3%), H-24 → L (6%), H-24 → L+2 (8%), H-24 → L+3 (2%), H-24 → L+18 (5%), H-9 → L (3%), H-9 → L+1 (5%), H-1 → L (7%), H → L+5 (4%) | 0.00 | ||||
| S0 → S19 | 151 | 8.19 | H-1 → L+5 (12%) H-24 → L (5%), H-24 → L+1 (3%), H-24 → L+2 (8%), H-24 → L+18 (5%), H-9 → L+1 (3%), H-4 → L+4 (6%), H-1 → L (4%), H-1 → L+1 (3%), H-1 → L+2 (3%), H → L+5 (2%), H → L+18 (8%) | 0.01 | ||||
| S0 → S20 | 150 | 8.26 | H-2 → L+6 (13%), H-15 → L+8 (6%), H-14 → L+13 (3%), H-13 → L+6 (4%), H-8 → L+6 (3%), H-7 → L+8 (5%), H-5 → L+11 (3%), H-5 → L+13 (3%), H-3 → L+6 (7%), H-3 → L+7 (2%), H-2 → L+7 (3%) | 0.01 | ||||
| *H: HOMO, L: LUMO | ||||||||
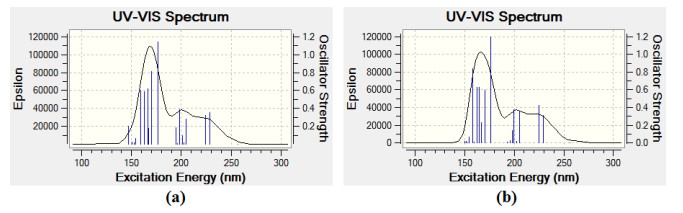
The analysis of calculated UV spectrum for the Vemurafenib drug shows λmax at 176 nm (f = 1.14) (Table 7). The charge transfer at λmax = 176 nm takes place at excited state S0 → S10 with nine electron configurations including H-3 → L+4 (35%), H-1 → L+2 (10%), H → L (13%), H-4 → L (3%), H-4 → L+2 (5%), H-1 → L+1 (7%), H → L+2 (6%), H → L+5 (5%) and H → L+6 (2%), in which the main transition is observed from HOMO-3 to LUMO+4 [H-3 → L+4 (35%)]. The excited state of S0 → S11 at 170 nm (f = 0.80) is also the other important excited state in the UV spectrum of Vemurafenib. The other excited states of Vemurafenib drug have very small intensity and do not play any role in the formation of electron spectrum of the title compound (Table 7). The theoretical electronic absorption spectrum of Vemurafenib drug in the solvent water is shown in Fig. 7(a).
After the adsorption of Vemurafenib drug on the BNNT(5,5-9), λmax appears at 177 nm (f = 1.19). The charge transfer at λmax = 177 takes place at excited state S0 → S10 and is defined by nine configurations including H-4 → L+4 (34%), H-1 → L+2 (12%), H → L (13%), H-9 → L (4%), H-9 → L+2 (5%), H-1 → L+1 (7%), H-1 → L+5 (2%), H → L+2 (6%), H → L+5 (5%) (Table 8). The major contribution to the absorption maxima is observed at HOMO-4 → LUMO+4 transition [H-4 → L+4], which contributes about 34% of the total excitations. The excited states of S0 → S16 and at 157 nm (f = 0.84) are also the other important excited states in the UV spectrum of complex BNNT(5,5-9)/Vemurafenib. The other excited states of the title compound have very small intensity (Table 8). Fig. 7b shows the calculated UV spectrum of complex BNNT/drug in the solvent water.
In the UV spectrum of the isolated Vemurafenib, λmax is observed at 176 nm, while after adsorption of the Vemurafenib over the BNNT it is enhanced to 177 nm. Thus, we found that adsorption of the Vemurafenib over the BNNT(5,5-9) nanotube change the value of λmax, although change is very partial.
In this research, the adsorption of Vemurafenib drug on BNNT(5,5-9) was studied at the M062/6-31G* level of theory. Some interesting outcomes of theoretical calculations are as follows:
(1) According to the obtained results, the adsorption process of Vemurafenib drug on BNNT(5,5-9) is an exothermic process and BNNT(5,5-9)/Vemurafenib is a stable complex.
(2) It is found that some geometrical parameters of Vemurafenib and BNNT are changed after adsorption process due to the formation of intermolecular non-bonded interaction.
(3) NBO analysis on a charge transfer from the molecule Vemurafenib to BNNT and from BNNT to Vemurafenib is predicted.
(4) The energy gaps between LUMO and HOMO in Vemurafenib increase after the adsorption of Vemurafenib on BNNT(5,5-9). This result indicates an increase in electrical conductivity of complex compared with the isolated drug and BNNT.
(5) The electronic properties, natural charges and chemical shift tensors are changed after interaction between Vemurafenib and BNNT(5,5-9).
(6) As a result, the quantum molecular descriptors change at adsorption process. The hardness and electronic chemical potential of the complex will be decreased, while electrophilicity and softness will be increased.
(7) Non-bonded interaction between the compound Vemurafenib and BNNT(5,5-9) partially changes the value of λmax.
We hope that our results of the adsorption properties of Vemurafenib on BNNT(5,5-9) can be used as in the adsorbent enhancing drugs delivery of the cancer cells and to support decreased drug interaction with healthy tissue.
Bertrand, N.; Leroux, J. C. The journey of a drug-carrier in the body: an anatomo-physiological perspective. J. Control. Release 2012, 161, 152–163. doi: 10.1016/j.jconrel.2011.09.098
Kumar, B.; Jalodia, K.; Kumar, P.; Gautam, H. K. Recent advances in nanoparticle-mediated drug delivery. J. Drug Deliv. Sci. Technol. 2017, 41, 260–268. doi: 10.1016/j.jddst.2017.07.019
Wang, Y.; Wang, F.; Shen, Y.; He, Q.; Guo, S. Tumor-specific disintegratable nanohybrids containing ultrasmall inorganic nanoparticles: from design and improved properties to cancer applications. Mater. Horiz. 2018, 5, 184–205. doi: 10.1039/C7MH01071K
Padma, V. D.; Jain, S. ed. Targeted Drug Delivery: Concepts and Design Advances in Delivery Science and Technology. Springer 2014.
Bae, Y. H.; Mrsny, R. J.; Park, K. ed. Cancer Targeted Drug Delivery: An Elusive Dream. Springer Science & Business Media 2013.
Dai, L.; Liu, J.; Luo, Z.; Li, M.; Cai, K. Tumor therapy: targeted drug delivery systems. J. Mater. Chem. B 2016, 4, 6758–6772. doi: 10.1039/C6TB01743F
Bhirde, A. A.; Patel, V.; Gavard, J.; Zhang, G.; Sousa, A. A.; Masedunskas, A.; Leapman, R. D.; Weigert, R.; Gutkind, J. S.; Rusling, J. F. Targeted killing of cancer cells in vivo and in vitro with EGF-directed carbon nanotubebased drug delivery. ACS Nano 2009, 3, 307–316. doi: 10.1021/nn800551s
Heister, E.; Neves, V.; Tîlmaciu, C.; Lipert, K.; Beltrán, V. S.; Coley, H. M.; Silva, S. R. P.; McFadden, J. Triplefunctionalisation of single-walled carbon nanotubes with doxorubicin, a monoclonal antibody, and a fluorescentmarker for targeted cancer therapy. Carbon 2009, 47, 2152–2160. doi: 10.1016/j.carbon.2009.03.057
Raju, H. B.; Goldberg, J. L. Nanotechnology for ocular therapeutics and tissue repair. Expert Rev. Ophthalom. 2008, 3, 431–436. doi: 10.1586/17469899.3.4.431
(a) https://www.curemelanoma.org (b)https://www.cancerresearchuk.org.
Zhi, C.; Bando, Y.; Tang, C.; Golberg, D. Boron nitride nanotubes. Mater. Sci. Eng. R Rep. 2010, 70, 92–111. doi: 10.1016/j.mser.2010.06.004
Chen, X.; Wu, P.; Rousseas, M.; Okawa, D.; Gartner, Z.; Zettl, A.; Bertozzi, C. R. Boron nitride nanotubesarenoncytotoxic and can be functionalized for interaction with proteins and cells. J. Am. Chem. Soc. 2009, 131, 890–891. doi: 10.1021/ja807334b
Chen, J.; Chen, S.; Zhao, X.; Kuznetsova, L. V.; Wong, S. S.; Ojima, I. Functionalized single-walled carbonnanotubes as rationally designed vehicles for tumor-targeted drug delivery. J. American Chem. Soc. 2008, 130, 16778–16785. doi: 10.1021/ja805570f
Han, W.; Bando, Y.; Kurashima, K.; Sato, T. Synthesis of boron nitride nanotubes from carbon nanotubes by a substitution reaction. Appl. Phys. Lett. 1998, 73, 3085–3087. doi: 10.1063/1.122680
Terrones, M.; Romo-Herrera, J.; Cruz-Silva, E.; López-Urías, F.; Munoz-Sandoval, E.; Velázquez-Salazar, J.; Terrones, H.; Bando, Y.; Golberg, D. Pure and doped boron nitride nanotubes. Mater. Today 2007, 10, 30–38.
Yang, C. K. Exploring the interaction between the boron nitride nanotube and biological molecules. Comput. Phys. Commun. 2011, 182, 39–42. doi: 10.1016/j.cpc.2010.07.040
Zhao, J. X.; Ding, Y. H. Theoretical investigation of the divacancies in boron nitride nanotubes: propertiesand surface reactivity toward various adsorbates. J. Chem. Phys. 2009, 131, 014706–014712. doi: 10.1063/1.3167409
(a) Chen, Y.; Zhou, J.; Campbell, S. J.; Caer, G. L. Boron nitride nanotubes: pronounced resistance to oxidation. Appl. Phys. Lett. 2004, 84, 2430–2432. (b) Cohen, M. L.; Zettl, A. The physics of boron nitride nanotubes. Phys. Today 2010, 63, 34–38.
Kalay, S.; Yilmaz, Z.; Sen, O.; Emanet, M.; Kazanc, E.; Çulha, M. Synthesis of boron nitride nanotubes and their applications. Beilstein. J. Nanotech. 2015, 6, 84–102. doi: 10.3762/bjnano.6.9
Mukhopadhyay, S.; Scheicher, R. H.; Pandey, R.; Karna, S. P. Sensitivity of boron nitride nanotubes toward biomolecules of different polarities. J. Phys. Chem. Lett. 2011, 2, 2442–2447. doi: 10.1021/jz2010557
Sheikhi, M.; Shahab, S.; Filippovich, L.; Khaleghian, M.; Dikusar, E.; Mashayekhi, M. Interaction between new synthesized derivative of (E, E)-azomethines and BN(6, 6-7) nanotube for medical applications: Geometry optimization, molecular structure, spectroscopic (NMR, UV/Vis, excited state), FMO, MEP and HOMO-LUMO investigations. J. Mol. Struct. 2017, 1146, 881–888. doi: 10.1016/j.molstruc.2017.06.017
Sheikhi, M.; Shahab, S.; Khaleghian, M.; Kumar, R. Interaction between new anti-cancer drug syndros and CNT(6, 6-6) nanotube for medical applications: geometry optimization, molecular structure, spectroscopic (NMR, UV/Vis, excited state), FMO, MEP and HOMO-LUMO investigation. Appl. Surf. Sci. 2018, 434, 504–513. doi: 10.1016/j.apsusc.2017.10.154
Sheikhi, M.; Shahab, S.; Khaleghian, M.; Haji Hajikolaee, F.; Balakhanava, I.; Alnajjar, R. Adsorption properties of the molecule resveratrol on CNT(8, 0-10) nanotube: Geometry optimization, molecular structure, spectroscopic (NMR, UV/Vis, excited state), FMO, MEP and HOMO-LUMO investigations. J. Mol. Struct. 2018, 1160, 479–487. doi: 10.1016/j.molstruc.2018.01.005
Sheikhi, M.; Shahab, S.; Alnajjar, R.; Ahmadianarog, M. Adsorption properties of the new anti-cancer drug alectinib on CNT(6, 6-6) nanotube: Geometry optimization, molecular structure, spectroscopic (NMR, UV/Vis, Excited State), FMO, MEP and HOMO-LUMO investigations. J. Clust. Sci. 2019, 30, 83–96. doi: 10.1007/s10876-018-1460-9
Shahab, S.; Filippovich, L.; Sheikhi, M.; Kumar, R.; Dikusar, E.; Yahyaei, H.; Muravsky, A. Polarization, excited states, trans-cis properties and anisotropy of thermal and electrical conductivity of the 4-(phenyldiazenyl)aniline in PVA matrix. J. Mol. Struct. 2017, 1141, 703–709. doi: 10.1016/j.molstruc.2017.04.014
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09 Revision A02, Gaussian, Inc., Wallingford CT 2009.
Frisch, A.; Nielsen, A. B.; Holder, A. J. Gauss View Users Manual, Gaussian Inc. 2000.
Sheikhi, M.; Shahab, S.; Filippovich, L.; Yahyaei, H.; Dikusar, E.; Khaleghian, M. New derivatives of (E, E)-azomethines: design, quantum chemical modeling, spectroscopic (FT-IR, UV/Vis, polarization) studies, synthesis and their applications: experimental and theoretical investigations. J. Mol. Struct. 2018, 1152, 368–385. doi: 10.1016/j.molstruc.2017.09.108
Shahab, S.; Sheikhi, M.; Filippovich, L.; DikusarAnatol'evich, E.; Yahyaei, H. Quantum chemical modeling of new derivatives of (E, E)-azomethines: synthesis, spectroscopic (FT-IR, UV/Vis, polarization) and thermophysical investigations. J. Mol. Struct. 2017, 1137, 335–348. doi: 10.1016/j.molstruc.2017.02.056
Weinhold, F.; Landis, C. R. Neutral bond orbitals and extensions of localized. Chem. Educ. Res. Pract. Eur. 2001, 2, 91–104. doi: 10.1039/B1RP90011K
Shahab, S.; Sheikhi, M.; Filippovich, L.; Khaleghian, M.; Dikusar, E.; Yahyaei, H.; Yousefzadeh Borzehandani, M. Spectroscopic studies (Geometry optimization, E → Z isomerization, UV/Vis, excited states, FT-IR, HOMO-LUMO, FMO, MEP, NBO, polarization) and anisotropy of thermal and electrical conductivityof new azomethine dyes in stretched polymer matrix. Silicon 2018, 10, 2361–2385. doi: 10.1007/s12633-018-9773-8
Sheikhi, M.; Shahab, S.; Filippovich, L.; Dikusar E.; Khaleghian, M. DFT investigations (Geometry optimization, UV/Vis, FT-IR, NMR, HOMO-LUMO, FMO, MEP, NBO, excited states) and the syntheses of new pyrimidine dyes. Chin. J. Struct. Chem. 2018, 37, 1201–1222.
Shahab, S.; Sheikhi, M.; Filippovich, L.; Dikusar, E.; Yahyaei, H.; Kumar, R.; Khaleghian, M. Design of geometry, synthesis, spectroscopic (FT-IR, UV/Vis, excited state, polarization) and anisotropy (thermal conductivity and electrical) properties of new synthesized derivatives of (E, E)-azomethines in colored stretched poly (vinyl alcohol) matrix. J. Mol. Struct. 2018, 1157, 536–550. doi: 10.1016/j.molstruc.2017.12.094

Figure 2 Various configurations of BNNT(5,5-9)/Vemurafenib complex optimized by the PM6 method

Figure 3 Optimized geometry of the molecules (a): Vemurafenib, (b): BNNT(5,5-9) and (c): complex BNNT(5,5-9)/Vemurafenib using M062X/6-31G* level of theory

Figure 4 Calculated HOMO and LUMO orbitals of the compounds Vemurafenib, BNNT(5,5-9), and complex BNNT(5,5-9)/Vemurafenib at the M062X/6-31G* level of theory

Figure 5 DOS plot of the compounds Vemurafenib, BNNT(5,5-9), and complex BNNT(5,5-9)/Vemurafenib at the M062X/6-31G* level of theory

Figure 7 UV spectra of the Vemurafenib drug (a) and complex BNNT(5,5-9)/Vemurafenib, (b) Calculated by TDM062X/6-31G* the method
Table 1. Energy Parameters for Various Configurations BNNT(5,5-9)/Vemurafenib Complex Optimized by the PM6 Method
| Systems | HF (Hartree) |
| State I | –3.6435114 |
| State II | –3.6471549 |
| State III | –3.6463678 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Thermochemical Parameters of the Molecules Vemurafenib, BNNT(5,5-9) and Complex BNNT(5,5-9)/Vemurafenib Calculated by the M062X/6-31G* Level of Theory
| Parameter | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib |
| E+G (Hartree) | –2334.2965 | –3199.1035 | 5534.6514 |
| E+H (Hartree) | –2334.2018 | –3198.9859 | 5534.3940 |
| E+T (Hartree) | –2334.2028 | –3198.9869 | 5534.5237 |
| E+ZPE (Hartree) | –2334.2318 | –3199.0351 | 5534.1382 |
| S (cal/mol·K) | 199.189 | 247.353 | 300.498 |
下载: 导出CSV
Table 3. Calculated Bond Lengths (Å) of the Molecule Vemurafenib, BNNT(5,5-9) and Complex BNNT(5,5-9)/Vemurafenib Using the M062X/6-31G* Method in a Solvent Water
| Bond length | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib |
| N(14)–C(15) | 1.355 | - | 1.354 |
| C(19)–C(24) | 1.391 | - | 1.387 |
| C(23)–C(24) | 1.391 | - | 1.392 |
| C(23)–N(25) | 1.426 | - | 1.427 |
| C(24)–F(32) | 1.337 | - | 1.339 |
| N(25)–S(26) | 1.696 | - | 1.687 |
| S(26)–O(27) | 1.462 | - | 1.462 |
| S(26)–O(28) | 1.459 | - | 1.463 |
| S(26)–C(29) | 1.873 | - | 1.782 |
| C(29)–C(30) | 1.527 | - | 1.527 |
| C(30)–C(31) | 1.526 | - | 1.526 |
| N(14)–H(40) | 1.011 | - | 1.014 |
| C(15)–H(41) | 1.081 | - | 1.083 |
| N(25)–H(44) | 1.021 | - | 1.020 |
| C(30)–H(47) | 1.094 | - | 1.095 |
| C(30)–H(48) | 1.094 | - | 1.096 |
| C(31)–H(49) | 1.092 | - | 1.091 |
| B(61)–N(63) | - | 1.453 | 1.452 |
| B(72)–N(73) | - | 1.420 | 1.428 |
| B(72)–N(74) | - | 1.453 | 1.450 |
| N(83)–B(84) | - | 1.445 | 1.442 |
| N(83)–B(93) | - | 1.420 | 1.419 |
| B(93)–N(95) | - | 1.453 | 1.454 |
| B(61)–N(131) | - | 1.420 | 1.420 |
| B(94)–N(105) | - | 1.445 | 1.445 |
| B(104)–N(105) | - | 1.420 | 1.419 |
| B(125)–N(115) | - | 1.420 | 1.420 |
| B(125)–N(127) | - | 1.453 | 1.453 |
| B(126)–N(131) | - | 1.445 | 1.446 |
| B(61)–H(134) | - | 1.195 | 1.196 |
| B(72)–H(137) | - | 1.195 | 1.192 |
| N(73)–H(138) | - | 1.012 | 1.015 |
| N(83)–H(140) | - | 1.012 | 1.016 |
| B(93)–H(142) | - | 1.195 | 1.196 |
| N(105)–H(146) | - | 1.012 | 1.012 |
| N(131)–H(151) | - | 1.012 | 1.012 |
下载: 导出CSV
Table 4. Donor-acceptor Interactions and the Second-order Perturbation Energies (E(2), kcal/mol) Related to Charge Transfer between Vemurafenib and BNNT(5,5-9) in Complex Calculated Using the M062X/6-31G* Method
| Donor (i) | Acceptor (j) | E(2)a (kcal/mol) | E(j)-E(i)b (a.u.) | F(i, j)c (a.u.) |
| π(C(11)–N(14)) | π*(B(72)–N(74)) | 0.32 | 0.49 | 0.011 |
| σ(N(14)–H(40)) | σ*(B(72)–N(73)) | 0.07 | 1.31 | 0.009 |
| π*(B(72)–N(74)) | 0.17 | 0.87 | 0.011 | |
| σ(C(15)–H(41)) | σ*(N(73)–H(138)) | 0.12 | 1.20 | 0.011 |
| σ(C(30)–H(48)) | π*(B(61)–N(63)) | 0.15 | 0.66 | 0.009 |
| σ(C(31)–H(49)) | σ*(N(115)–H(148)) | 0.27 | 1.12 | 0.016 |
| σ*(B(125)–N(127)) | 0.09 | 1.08 | 0.009 | |
| π*(B(125)–N(127)) | 0.17 | 0.66 | 0.010 | |
| n1(O(27)) | σ*(N(83) H(140)) | 3.18 | 1.46 | 0.061 |
| σ*(B(93)–N(95)) | 0.10 | 1.41 | 0.011 | |
| n2(O(27)) | σ*(B(72)–H(137)) | 0.17 | 0.96 | 0.012 |
| σ*(N(83)–H(140)) | 0.23 | 0.94 | 0.014 | |
| n1(O(28)) | σ*(B(72)–N(73)) | 0.05 | 1.42 | 0.008 |
| σ*(B(72)–N(74)) | 0.22 | 1.41 | 0.016 | |
| n2(O(28)) | σ*(N(73)–H(138)) | 3.29 | 1.45 | 0.062 |
| σ*(B(72)–N(74)) | 0.11 | 0.90 | 0.009 | |
| π*(C(11)–N(14)) | π*(B(72)–N(74)) | 0.38 | 0.13 | 0.009 |
| π*(C(15)–C(16)) | π*(B(72)–N(74)) | 0.47 | 0.04 | 0.008 |
| σ*(S(26)–O(27)) | σ*(N(83)–H(140)) | 0.76 | 0.24 | 0.042 |
| σ*(S(26)–O(28)) | σ*(N(73)–H(138)) | 0.76 | 0.23 | 0.040 |
| π(B(61)–N(63)) | σ*(N(14)–H(40)) | 0.09 | 0.85 | 0.008 |
| σ(B(61)–H(134)) | σ*(C(30)–H(48)) | 0.29 | 1.01 | 0.015 |
| π(B(62)–N(73)) | σ*(C(11)–N(14)) | 0.14 | 0.84 | 0.010 |
| σ*(N(14)–H(40)) | 2.95 | 0.87 | 0.047 | |
| σ*(C(15)–C(16)) | 0.09 | 0.96 | 0.009 | |
| σ*(C(15)–H(41)) | 0.09 | 0.88 | 0.008 | |
| π(B(72)–N(73)) | σ*(N(14)–H(40)) | 0.09 | 1.30 | 0.009 |
| σ(B(72)–H(137)) | π*(C(15)–C(16)) | 0.11 | 0.52 | 0.007 |
| σ*(S(26)–O(27)) | 0.18 | 0.80 | 0.011 | |
| σ*(S(26)–O(28)) | 0.15 | 0.80 | 0.010 | |
| σ*(S(26)–C(29)) | 0.11 | 0.67 | 0.008 | |
| σ(N(73)–H(138)) | σ*(C(15)–H(41)) | 0.12 | 1.24 | 0.011 |
| σ*(S(26)–O(28)) | 0.10 | 1.02 | 0.009 | |
| σ(N(83)–B(84)) | σ*(S(26)–O(27)) | 0.06 | 1.10 | 0.008 |
| σ(N(83)–H(140)) | σ*(S(26)–C(27)) | 0.09 | 1.02 | 0.009 |
| π(N(115)–B(116)) | σ*(C(31)–H(49)) | 0.16 | 1.32 | 0.013 |
| σ*(C(31)–H(49)) | 0.10 | 0.88 | 0.009 | |
| σ(N(115)–H(148)) | σ*(C(31)–H(49)) | 0.18 | 1.24 | 0.013 |
| σ(B(125)–H(150)) | σ*(C(31)–H(49)) | 0.48 | 1.03 | 0.020 |
| π*(B(61)–N(63)) | σ*(C(30)–H(48)) | 0.30 | 0.44 | 0.030 |
| π*(B(72)–N(74)) | σ*(N(25)–S(26)) | 0.15 | 0.07 | 0.006 |
| π*(B(93)–N(95)) | σ*(C(30)–H(47)) | 0.06 | 0.43 | 0.013 |
| π*(B(125)–N(127)) | σ*(C(31)–H(49)) | 0.19 | 0.46 | 0.025 |
| a E(2) Energy of hyperconjucative interactions. b Energy difference between donor and acceptor i and j NBO orbitals. c F(i, j) is the Fock matrix element between i and j NBO orbitals. | ||||
下载: 导出CSV
Table 5. Calculated Electronic Properties of the Vemurafenib Drug, BNNT(5,5-9) and the Complex BNNT(5,5-9)/Vemurafenib at the M062X/6-31G* Level of Theory in the Solvent Water
| Property | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib |
| Dipole moment (Debye) | 6.78 | 0.00 | 11.62 |
| E(Hartree) | –2334.61294 | –3199.69538 | –5534.32742 |
| Ead (Hartree) | – | – | –0.0191 |
| EHOMO (eV) | –7.66 | –7.95 | –7.53 |
| ELUMO (eV) | –1.06 | 0.94 | –0.89 |
| Eg (eV) | 6.60 | 7.01 | 6.64 |
| I (eV) | 7.66 | 7.95 | 7.53 |
| A (eV) | 1.06 | 0.94 | 0.89 |
| χ (eV) | 4.36 | 4.44 | 4.21 |
| η (eV) | 3.30 | 3.50 | 3.32 |
| μ (eV) | –4.63 | –4.44 | –4.21 |
| ω (eV) | 3.24 | 2.81 | 2.66 |
| S (eV) | 0.15 | 0.14 | 0.15 |
下载: 导出CSV
Table 6. Natural Charges (NBO Charges, e) and NMR Parameters (ppm) Including CSI and CSA for the Selected Atoms in the Molecules Vemurafenib, BNNT(5,5-9) and the Complex BNNT(5,5-9)/Vemurafenib Calculated by the M062X/6-31G* Method
| Atoms | Vemurafenib | BNNT(5,5-9) | BNNT(5,5-9)/Vemurafenib | ||||||||
| Charge | CSI | CSA | Charge | CSI | CSA | Charge | CSI | CSA | |||
| N(14) | –0.557 | 105.44 | 116.82 | - | - | - | –0.558 | 98.26 | 126.29 | ||
| C(15) | 0.072 | 49.29 | 152.43 | - | - | - | 0.071 | 46.69 | 155.36 | ||
| C(23) | 0.038 | 66.62 | 128.36 | - | - | - | 0.040 | 65.24 | 126.80 | ||
| C(24) | 0.464 | 30.91 | 113.82 | - | - | - | 0.459 | 26.97 | 121.88 | ||
| N(25) | –0.916 | 137.17 | 143.47 | - | - | - | –0.936 | 147.43 | 155.12 | ||
| S(26) | 2.384 | 214.42 | 188.03 | - | - | - | 2.402 | 212.77 | 135.97 | ||
| O(27) | –0.994 | 118.02 | 98.47 | - | - | - | –0.999 | 120.24 | 69.14 | ||
| O(28) | –0.995 | 148.59 | 149.93 | - | - | - | –1.004 | 129.10 | 72.88 | ||
| C(29) | –0.719 | 135.95 | 68.85 | - | - | - | –0.710 | 132.74 | 68.67 | ||
| C(30) | –0.490 | 179.28 | 10.25 | - | - | - | –0.489 | 177.77 | 10.66 | ||
| C(31) | –0.691 | 182.49 | 21.56 | - | - | - | –0.683 | 180.39 | 19.84 | ||
| F(32) | –0.335 | 312.65 | 177.15 | - | - | - | –0.330 | 289.69 | 170.36 | ||
| F(33) | –0.330 | 295.90 | 146.47 | - | - | - | –0.331 | 296.90 | 167.99 | ||
| H(40) | 0.476 | 23.61 | 9.61 | - | - | - | 0.476 | 23.25 | 15.82 | ||
| H(41) | 0.271 | 23.97 | 8.28 | - | - | - | 0.267 | 23.11 | 9.95 | ||
| H(44) | 0.452 | 26.82 | 8.00 | - | - | - | 0.465 | 26.80 | 8.76 | ||
| H(47) | 0.257 | 30.36 | 6.61 | - | - | - | 0.255 | 30.58 | 7.28 | ||
| H(48) | 0.255 | 30.40 | 6.80 | - | - | - | 0.250 | 30.69 | 8.58 | ||
| H(49) | 0.250 | 30.89 | 11.43 | - | - | - | 0.237 | 30.65 | 12.24 | ||
| B(61) | - | - | - | 0.818 | 78.57 | 59.03 | 0.813 | 79.02 | 58.06 | ||
| B(62) | - | - | - | 1.193 | 83.03 | 37.51 | 1.193 | 82.63 | 37.42 | ||
| N(63) | - | - | - | –1.217 | 134.42 | 208.98 | –1.216 | 134.53 | 209.64 | ||
| B(72) | - | - | - | 0.818 | 78.62 | 59.67 | 0.818 | 75.55 | 65.25 | ||
| N(73) | - | - | - | –1.147 | 144.41 | 148.88 | –1.177 | 142.77 | 150.78 | ||
| N(74) | - | - | - | –1.217 | 134.67 | 210.10 | –0.219 | 133.42 | 208.04 | ||
| N(83) | - | - | - | –1.147 | 144.30 | 149.41 | –0.152 | 141.05 | 154.80 | ||
| B(93) | - | - | - | 0.818 | 78.51 | 58.77 | 0.818 | 78.39 | 59.17 | ||
| B(94) | - | - | - | 1.194 | 83.03 | 37.85 | 1.193 | 82.82 | 37.68 | ||
| B(104) | - | - | - | 0.818 | 78.68 | 58.80 | 0.818 | 79.03 | 60.05 | ||
| N(105) | - | - | - | –1.147 | 144.31 | 149.54 | –1.147 | 143.88 | 151.17 | ||
| N(115) | - | - | - | –1.147 | 144.45 | 148.84 | –1.148 | 144.03 | 149.86 | ||
| B(125) | - | - | - | 0.818 | 78.13 | 58.63 | 0.813 | 78.23 | 60.97 | ||
| B(126) | - | - | - | 1.193 | 82.97 | 37.63 | 1.192 | 83.43 | 36.81 | ||
| N(127) | - | - | - | –1.217 | 134.56 | 209.54 | –1.217 | 135.68 | 206.23 | ||
下载: 导出CSV
Table 7. Electronic Absorption Spectrum of the Vemurafenib Calculated by TDM062X/6-31G* Method
| Excited state | Wavelength (nm) |
Excitation energy (eV) | Configurations composition (corresponding transition orbitals) |
Oscillator strength (f) | ||||
| S0 → S1 | 256 | 4.82 | H-9 → L (31%), H-9 → L+5 (14%), H-6 → L (18%) H-9 → L+1 (3%), H-9 → L+12 (2%), H-8 → L (2%), H-6 → L+5 (7%) |
0.00 | ||||
| S0 → S2 | 228 | 5.41 | H-1 → L (25%), H → L (15%), H → L+1 (14%), H → L+2 (17%) H-4 → L (2%), H-4 → L+6 (3%), H-3 → L+4 (2%), H-1 → L+1 (8%), H-1 → L+2 (2%) |
0.35 | ||||
| S0 → S3 | 224 | 5.53 | H-1 → L (25%), H → LU (24%), H → L+1 (24%), H-4 → L+2 (3%), H-1 → L+1 (3%), H-1 → L+6 (2%), H → L+2 (7%) |
0.32 | ||||
| S0 → S4 | 205 | 6.04 | H-1 → L (13%), H → L+2 (26%) H-7 → L (3%), H-5 → L (3%), H-4 → L (4%), H-4 → L+1 (4%), H-3 → L+4 (7%), H-1 → L+1 (4%), H-1 → L+2 (7%), H-1 → L+5 (6%), H → L+4 (2%) |
0.28 | ||||
| S0 → S5 | 202 | 6.11 | H-4 → L+4 (11%), H-3 → L+2 (19%), H → L+4 (35%), H-3 → L (6%), H-3 → L+1 (9%), H-3 → L+6 (5%), H-1 → L+4 (6%) |
0.01 | ||||
| S0 → S6 | 201 | 6.15 | H-5 → L+3 (19%), H-2 → L (35%), H-2 → L+1 (25%) H-2 → L+5 (9%) |
0.09 | ||||
| S0 → S7 | 198 | 6.24 | H-1 → L+1 (12%), H-1 → L+2 (24%), H-5 → L (6%), H-5 → L+1 (7%), H-4 → L (5%), H-3 → L+4 (5%), H-2 → L+3 (8%), H-1 → L (2%), H-1 → L+5 (5%), H → L+1 (4%), H → L+5 (2%), H → L+6 (5%) | 0.39 | ||||
| S0 → S8 | 195 | 6.32 | H-6 → L+1 (22%), H-6 → L+2 (19%), H-9 → L (3%), H-9 → L+1 (5%), H-9 → L+2 (5%), H-6 → L (8%), H-6 → L+5 (7%), H-2 → L+3 (2%) | 0.00 | ||||
| S0 → S9 | 194 | 6.36 | H-5 → L (16%), H-2 → L+3 (29%), H-5 → L+1 (9%), H-5 → L+5 (2%), H-1 → L (5%), H-1 → L+1 (5%), H-1 → L+2 (5%), H-1 → L+5 (7%) | 0.18 | ||||
| S0 → S10 | 176 | 7.03 | H-3 → L+4 (35%), H-1 → L+2 (10%), H → L (13%), H-4 → L (3%), H-4 → L+2 (5%), H-1 → L+1 (7%), H → L+2 (6%), H → L+5 (5%), H → L+6 (2%) | 1.14 | ||||
| S0 → S11 | 170 | 7.27 | H-1 → L+1 (20%), H-1 → L+2 (12%), H-1 → L+6 (13%), H → L+2 (10%), H-7 → L(4%), H-5 → L (3%), H-3 → L+2 (3%), H-3 → L+4 (5%), H-2 → L+3 (5%), H-1 → L (3%), H-1 → L+5 (3%), H → L+1 (2%) | 0.80 | ||||
| S0 → S12 | 167 | 7.41 | H-3 → L+4 (12%), H → L (18%), H → L+1 (15%), H → L+2 (12%), H → L+5(13%), H-4 → L (6%), H-4 → L+2 (3%), H-4 → L+5 (3%), H → L+6 (5%) |
0.17 | ||||
| S0 → S13 | 166 | 7.43 | H-5 → LUMO (28%), H-2 → L+3 (37%), H-5 → L+1 (5%), H-1 → L+2 (2%) | 0.61 | ||||
| S0 → S14 | 162 | 7.61 | H-3 → L (10%), H-3 → L+1 (13%), H-3 → L+2 (19%), H → L+4 (30%), H-4 → L+4 (3%), H-3 → L+6 (3%), H-1 → L+1 (3%), H-1 → L+4 (3%) | 0.58 | ||||
| S0 → S15 | 159 | 7.78 | H-4 → L+7 (30%), H → L+7 (43%), H-13 → L+7 (5%), H-7 → L+7 (2%), H-1 → L+7 (8%) | 0.00 | ||||
| S0 → S16 | 158 | 7.80 | H-5 → L+3 (60%), H-2 → L (20%), H-5 → L+5 (3%), H-4 → L+3 (3%), H-2 → L+1 (4%) |
0.60 | ||||
| S0 → S17 | 154 | 8.02 | H-4 → L+2 (16%), H-3 → L+4 (20%), H → L+6 (10%), H-7 → L (3%), H-7 → L+1 (6%), H-6 → L (2%), H-6 → L+2 (2%), H-6 → L+6 (2%), H-4 → L (3%), H-4 → L+1 (3%), H-1 → L+1 (7%), H-1 → L+5 (2%), H-1 → L+6 (5%) | 0.06 | ||||
| S0 → S18 | 152 | 8.13 | H-6 → L+2 (16%), H-6 → L+6 (10%), H-9 → L (2%), H-9 → L+2 (4%), H-9 → L+6 (2%), H-7 → L+1 (4%), H-6 → L (7%), H-4 → L (2%), H-4 → L+1 (3%), H-1 → L (5%), H-1 → L+1 (9%), H-1 → L+5 (8%), H → L+5 (3%) | 0.01 | ||||
| S0 → S19 | 150 | 8.21 | H-1 → L+5 (20%), H → L+6 (10%), H-6 → L (5%), H-6 → L+2 (8%), H-6 → L+6 (6%), H-4 → L+1 (4%), H-4 → L+2 (3%), H-3 → L+4 (5%), H-1 → L (6%), H-1 → L+1 (7%), H-1 → L+2 (3%), H → L+5 (2%) | 0.02 | ||||
| S0 → S20 | 147 | 8.42 | H-5 → L+5 (28%), H-5 → L+1 (9%), H-5 → L+3 (2%), H-4 → L (3%), H-4 → L+1 (6%), H-2 → L+1 (3%), H-2 → L+3 (7%), H-2 → L+5 (9%), H-1 → L+5 (2%), H → L (3%) | 0.19 | ||||
| *H: HOMO, L: LUMO | ||||||||
下载: 导出CSV
Table 8. Electronic Absorption Spectrum of Complex BNNT(5,5-9)/Vemurafenib Calculated by the TDM062X/6-31G* Method
| Excited state | Wavelength (nm) |
Excitation energy (eV) | Configurations composition (corresponding transition orbitals) |
Oscillator strength (f) | ||||
| S0 → S1 | 256 | 4.84 | H-36 → L (12%), H-24 → L (19%), H-41 → L (3%), H-36 → L+5 (5%), H-35 → L (3%), H-33 → L (6%), H-33 → L+5 (3%), H-32 → L (4%), H-24 → L+5 (8%) | 0.00 | ||||
| S0 → S2 | 228 | 5.41 | H-1 → L (30%), H-1 → L+1 (10%), H → L (10%), H → L+1 (12%), H → L+2 (12%), H-9 → L (3%), H-4 → L+4 (2%), H-1 → L+2 (2%), H → L+3 (5%) | 0.30 | ||||
| S0 → S3 | 224 | 5.51 | H-1 → L (19%), H → L (28%), H → L+1 (26%), H-9 → L+2 (4%), H-1 → L+5 (2%), H → L+2 (8%) | 0.42 | ||||
| S0 → S4 | 205 | 6.03 | H-1 → LUMO (14%), H → L+2 (23%), H-26 → L (4%), H-9 → L (5%), H-9 → L+1 (4%), H-4 → L+4 (6%), H-1 → L+1 (6%), H-1 → L+2 (3%), H-1 → L+3 (4%), H-1 → L+5 (7%), H → L (2%), H → L+3 (3%), H → L+5 (2%) | 0.35 | ||||
| S0 → S5 | 202 | 6.11 | H-9 → L+4 (12%), H-4 → L+1 (10%), H-4 → L+2 (12%), H → L+4 (34%), H-4 → L (6%), H-4 → L+3 (6%), H-4 → L+18 (3%), H-1 → L+4 (8%) | 0.01 | ||||
| S0 → S6 | 199 | 6.22 | H-1 → L+2 (28%), H-12 → L+3 (3%), H-9 → L (3%), H-9 → L+1 (4%), H-6 → L (6%), H-6 → L+1 (5%), H-4 → L+4 (6%), H-1 → L+1 (8%), H-1 → L+5 (3%), H → L+1 (4%), H → L+5 (2%), H → L+18 (4%) | 0.37 | ||||
| S0 → S7 | 198 | 6.24 | H-12 → L+3 (14%), H-6 → L (28%), H-6 → L+1 (20%), H-12 → L+2 (4%), H-6 → L+5 (7%), H-1 → L+1 (2%), H-1 → L+2 (4%) | 0.14 | ||||
| S0 → S8 | 196 | 6.32 | H-24 → L+1 (18%), H-24 → L+2 (20%), H-33 → L+1 (2%), H-33 → L+2 (3%), H-32 → L+2 (2%), H-24 → L (7%), H-24 → L+5 (6%) | 0.02 | ||||
| S0 → S9 | 192 | 6.43 | H-12 → L (23%), H-12 → L+1 (14%), H-6 → L+3 (27%), H-13 → L (3%), H-13 → L+1 (2%), H-12 → L+5 (3%), H-6 → L+2 (6%), H-1 → L (2%), H-1 → L+5 (3%) | 0.01 | ||||
| S0 → S10 | 177 | 7.02 | H-4 → L+4 (34%), H-1 → L+2 (12%), H → L (13%), H-9 → L (4%), H-9 → L+2 (5%), H-1 → L+1 (7%), H-1 → L+5 (2%), H → L+2 (6%), H → L+5 (5%) | 1.19 | ||||
| S0 → S11 | 170 | 7.26 | H-1 → L+1 (23%), H → L+2 (13%), H-26 → L (4%), H-4 → L+4 (7%), H-1 → L (4%), H-1 → L+2 (7%), H-1 → L+3 (6%), H-1 → L+5 (3%), H-1 → L+18 (9%) | 0.59 | ||||
| S0 → S12 | 167 | 7.41 | H-4 → L+4 (12%), H → L (18%), H → L+1 (15%), H → L+5 (12%) H-9 → L (6%), H-9 → L+5 (3%), H → L+2 (5%), H → L+3 (8%), H → L+18 (3%) | 0.23 | ||||
| S0 → S13 | 164 | 7.53 | H-12 → L (26%), H-6 → L+3 (27%), H-13 → L (4%), H-12 → L+1 (3%), H-12 → L+3 (2%), H-6 → L+2 (6%), H-4 → L (2%), H-4 → L+1 (3%), H-4 → L+2 (3%), H → L+4 (5%) | 0.62 | ||||
| S0 → S14 | 162 | 7.61 | H-4 → L+1 (13%), H-4 → L+2 (11%), H → L+4 (25%), H-12 → L (5%), H-9 → L+4 (3%), H-6 → L+3 (5%), H-4 → L (9%), H-4 → L+3 (6%), H-1 → L+1 (3%), H-1 → L+4 (4%) | 0.63 | ||||
| S0 → S15 | 159 | 7.78 | H-9 → L+23 (32%), H-1 → L+23 (10%), H → L+23 (39%), H-48 → L+23 (5%), H-26 → L+23 (2%) | 0.00 | ||||
| S0 → S16 | 157 | 7.85 | H-12 → L+2 (10%), H-12 → L+3 (40%), H-6 → L (20%), H-13 → L+3 (6%), H-12 → L+5 (2%), H-6 → L+1 (5%), H-6 → L+3 (2%), H-1 → L+3 (2%) | 0.84 | ||||
| S0 → S17 | 154 | 8.01 | H-9 → L+2 (11%), H-4 → L+4 (19%), H-26 → L (3%), H-26 → L+1 (7%), H-9 → L (3%), H-9 → L+1 (2%), H-9 → L+3 (4%), H-1 → L+1 (8%), H-1 → L+5 (4%), H-1 → L+18 (4%), H → L+18 (5%) | 0.06 | ||||
| S0 → S18 | 153 | 8.10 | H-1 → L+1 (10%), H-1 → L+5 (13%), H-26 → L+1 (3%), H-24 → L (6%), H-24 → L+2 (8%), H-24 → L+3 (2%), H-24 → L+18 (5%), H-9 → L (3%), H-9 → L+1 (5%), H-1 → L (7%), H → L+5 (4%) | 0.00 | ||||
| S0 → S19 | 151 | 8.19 | H-1 → L+5 (12%) H-24 → L (5%), H-24 → L+1 (3%), H-24 → L+2 (8%), H-24 → L+18 (5%), H-9 → L+1 (3%), H-4 → L+4 (6%), H-1 → L (4%), H-1 → L+1 (3%), H-1 → L+2 (3%), H → L+5 (2%), H → L+18 (8%) | 0.01 | ||||
| S0 → S20 | 150 | 8.26 | H-2 → L+6 (13%), H-15 → L+8 (6%), H-14 → L+13 (3%), H-13 → L+6 (4%), H-8 → L+6 (3%), H-7 → L+8 (5%), H-5 → L+11 (3%), H-5 → L+13 (3%), H-3 → L+6 (7%), H-3 → L+7 (2%), H-2 → L+7 (3%) | 0.01 | ||||
| *H: HOMO, L: LUMO | ||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

