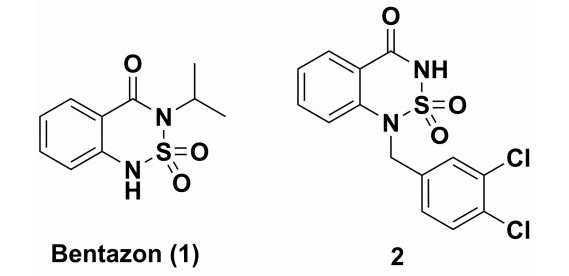
Figure 1.
Structures of bentazon (1) and PDE 7 inhibitor (2)
Synthesis, Crystal Structure and DFT Study of 6-Hydroxybentazon
Qiu-Yan ZHANG , Zhu-Yan HUANG , Yi LE , Xue ZOU , Zhi-Xu ZHOU , Chun-Shen ZHAO

Bentazon (1) has occupied important position in the herbicide market because it is a post-emergence herbicide with outstanding features of high efficiency, low toxicity, widespread applicability and good mixing properties, used for the selective control of broadleaf weeds and sedges in beans, rice, corn, peanuts, mint, etc[1-4]. Past researches have revealed bentazon derivatives (BTDs) have important physiological effects and serve as heterocyclic inhibitors of phosphodiesterase[5, 6]. For example, compound 2 (Fig. 1) is an inhibitor of phosphodiesterase (PDE) 7[5]. 6-Hydroxybentazon (11), the hydrolyzate of bentazon, has excellent synthetic utility as an intermediate in the preparation of BTDs because bentazon can be readily derived by introducing hydroxyl groups onto the benzene ring. It has a certain promoting effect on the development and application of bentazon in pesticides.
The bentazon of the backbone has been reported by Xuan Qi et al[7], and our team has published a patent report on the synthesis of 6-hydroxybenzon[8], but the crystal structure and DFT studies of 6-hydroxybenzon have never been reported. A synthesis method of 6-hydroxybentazon from 5-chloro-2-nitrobenzoic acid was chosen and designed in the paper based on the synthesis route of bentazon (Scheme 1). In addition, the crystal structure of 6-hydroxybentazon was confirmed by single-crystal X-ray diffraction analysis. The optimized structures of the molecules were calculated by density functional theory (DFT) using the B3LYP method with the 6-311G(d, p) basis set. Moreover, conformational analyses of the title compound were conducted and the structures optimized by the DFT method were compared with the X-ray structures. The results showed that the crystal structures determined by single-crystal X-ray diffraction fraction and DFT calculation were extremely close.
Mass spectra (MS) were taken in ESI mode on Agilent 1100 LC-MS (Agilent Technologies, Palo Alto, CA, U.S.A.). 1H NMR and 13C NMR spectra were recorded on a Bruker ARX-400, 400 MHz spectrometer (Bruker Bioscience, Billerica, MA, USA) with TMS as an internal standard. The IR spectrum of the title compound was recorded in the region of 4000~400 cm−1 using KBr pellet technique with 1.0 cm−1 resolution on a Bruker IFS-55V IR spectrometer (Bruker, Germany). The X-ray diffraction data of crystal 11 were recorded with a Bruker P4 X-diffractometer; the data were collected by using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 K. For 11, data collection: APEX2; cell refinement: SAINT; program used to solve structure: SHELXS-97; program used to refine structure and draw molecular figures: SHELXTL-97; program used to measure centroid-centroid distance: Mercury 2.3. All materials were obtained from commercial suppliers and used without further purification.
A mixture of 5-chloro-2-nitrobenzoic acid (30.0 g, 0.149 mol) and sodium hydroxide (47.4 g, 1.185 mol) in water (300 mL) was refluxed for 48 h. After cooling, the reaction mixture was poured into 3 mol/L hydrochloric acid (400 mL) to afford solid. Then, it was filtered and leached with water (100 mL), and the solvent was dried to obtain a light yellow powder (26.2 g, 96.2%).
A mixture of 5-hydroxy-2-nitrobenzoic acid (25.0 g, 0.137 mol) and concentrated sulfuric acid (14.9 mL, 0.274 mol) in methanol (200 mL) was refluxed for 6 h. After cooling, the reaction mixture was poured into ice water (200 mL) to afford solid. Then, it was filtered and leached with water (100 mL), and the solvent was dried to obtain a light yellow powder (26.3 g, 97.4%).
A mixture of methyl 5-hydroxy-2-nitrobenzoate (25.0 g, 0.127 mol), benzyl bromide (21.7 g, 0.127 mol) and potassium carbonate (43.8 g, 0.318 mol) in N, N-dimethylformamide (200 mL) was stirred at room temperature for 3 h. The reaction mixture was poured into water (200 mL) to afford solid. Then, it was filtered and leached with water (100 mL), and the solvent was dried to obtain a yellow powder (34.7 g, 95.3%).
A mixture of methyl 5-(benzyloxy)-2-nitrobenzoate (34.0 g, 0.118 mol), iron powder (13.2 g, 0.236 mol) and ammonium chloride (18.9 g, 0.354 mol) in EtOH/H2O (1:1) (200 mL) was refluxed for 12 h. After cooling, the reaction mixture was filtered and extracted with ethyl acetate (200 mL × 3). Then the organic phase was dehydrated with anhydrous sodium sulfate and concentrated at reduced pressure to obtain a white powder (26.0 g, 85.7%). 1H NMR (400 MHz, Chloroform-d) δ 7.48~7.29 (m, 6H), 7.02 (dd, J = 8.9, 3.0 Hz, 1H), 6.63 (d, J = 8.9 Hz, 1H), 5.44 (s, 2H), 4.99 (s, 2H), 3.87 (s, 3H).
A solution of isopropylamine (17.2 g, 0.291 mol) in dichloromethane was added dropwise chlorosulfonic acid (11.3 g, 0.097 mol) diluted with 20 mL dichloromethane under stirring at 0 ℃. The mixture was stirred for 1 h and dried under vacuum. And then phosphorus pentachloride (30.4 g, 0.146 mol) was added to the solution of residue in 250 mL toluene. After refluxing for 1 h at 110 ℃, the reaction solution was filtrated to remove inorganic substance and concentrated at reduced pressure to get the isopropylsulfamoyl chloride (8).
A mixture of methyl 2-amino-5-(benzyloxy)benzoate (25.0 g, 0.097 mol), triethylamine (14.8 g, 0.146 mol) and isopropylsulfamoyl chloride in toluene (200 mL) was stirred at room temperature for 3 h. The reaction mixture was poured into water (200 mL) and extracted with ethyl acetate (200 mL × 3). The organic phase was dehydrated with anhydrous sodium sulfate and concentrated at reduced pressure to obtain a white powder (36.7 g, 88.6%).
A mixture of methyl 5-(benzyloxy)-2-((N-isopropylsulfamoyl)amino)benzoate (36.0 g, 0.095 mol) and sodium methoxide (15.4 g, 0.285 mol) in methanol (300 mL) was stirred at 40 ℃ for 6 h. After cooling, the reaction mixture was poured into water (200 mL) and extracted with ethyl acetate (200 mL × 3). The organic phase was dehydrated with anhydrous sodium sulfate and concentrated at reduced pressure to obtain a white powder (28.8 g, 87.5%). 1H NMR (400 MHz, chloroform-d) δ 7.72 (d, J = 2.9 Hz, 1H), 7.47~7.32 (m, 5H), 7.21 (dd, J = 8.7, 2.9 Hz, 1H), 7.10 (d, J = 8.7 Hz, 1H), 5.12 (s, 2H), 5.01 (m, J = 7.0 Hz, 1H), 1.58 (d, J = 7.0 Hz, 6H).
Boron tribromide (71.4 g, 0.285 mol) was added to a solution of 6-(benzyloxy)-3-isopropyl-1H-benzo[c][1,2,6]thiadiazin-4(3H)-one 2, 2-dioxide (28.0 g, 0.095 mol) in dichloromethane (300 mL) at –78 oC and the solution was stirred for 30 min. Then, the mixture was stirred at room temperature for 1 h. The reaction mixture was poured into water (200 mL), filtered and extracted with dichloromethane (200 mL × 3). The organic phase was dehydrated with anhydrous sodium sulfate and concentrated at reduced pressure, and the residue was crystallized from chloroform to afford a white powder (22.2 g, 91.3%). IR (KBr pellets): ν 3381 (56%, Ar-OH), 3166, 1616 (58%, 66%, NH), 1219, 883, 834 (58%, 72%, 75%, ArH), 2995, 2948, 1465, 1404(73%, 78%, 47%, 70%, CH3), 1677(38%, C=O), 1503 (69%, C=C), 1315, 1161 (46%, 47%, SO2) cm-1. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 7.32 (d, J = 2.5 Hz, 1H), 7.08~6.97 (m, 2H), 4.80 (dt, J = 13.5, 6.7 Hz, 1H), 1.42 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 162.52, 155.37, 129.25, 123.45, 123.16, 121.71, 114.53, 48.14, 20.89. MS (ESI): m/z = 376.9 [M-H]-.
The crystal of the title compound 11 was cultivated from ethyl acetate and the colorless prism with dimensions of 0.22mm × 0.20mm × 0.18mm was selected for X-ray diffraction analysis. All measurements were made on a Bruker APEX Ⅱ diffractometer with Mo-Kα radiation (λ = 0.71073 Å). The data were collected at 296(2) K. The crystal is of orthorhombic system, space group Pbca with a = 9.5859(11), b = 12.8787(14), c = 14.3520(17) Å, V = 1657.1(3) Å3, Z = 6, density (calculated) = 1.541 g/cm3, and linear absorption coefficient is 0.298 mm-1. In the range of 1.48≤θ≤25.04º, a total of 10868 reflections were collected with 5845 unique ones (Rint = 0.0226), of which 4275 were observed with I > 2σ(I). The structure was solved by direct methods with SHELXS-97 program[9]. Refinements were done by full-matrix least-squares on F2 with SHELXL-97[9]. All of the non-H atoms were refined anisotropically by full-matrix leastsquares to give the final R = 0.0562 and wR = 0.1529 (w = 1/[σ2(Fo2) + (0.0997P)2 + 0.2530P], where P = (Fo2 + 2Fc2)/3), S = 1.039, (Δ/σ)max = 0.000, (Δρ)max = 0.693 and (Δρ)min = –0.472 e/Å3 by using the SHELXL program. The hydrogen atoms were placed in the calculated positions and refined isotropically.
The conformation of a molecule critically influences its physical and chemical properties[10-12]. Thus, reliable conformational analysis plays a key role in understanding the molecular structure. Initial conformational search for compound 11 was performed by the Spartan 08 program[13] with a molecular mechanics force field (MMFF)[14, 15]. Then, geometry optimizations and frequency calculation of all possible conformers were performed by using DFT/B3LYP/6-311G**[16, 17] in the Gaussian 09 package[18]. From the relative free energies, the percentage population of each conformation in a room-temperature equilibrium mixture can be predicted. The relative Gibbs free energies and Boltzmann distribution of 11 are shown in Table 1, and the conformers of 11 are presented in Fig. S1.
 DownLoad:
CSV
DownLoad:
CSV
| Conformer | G(kcal mol-1) | △G(kcal mol-1) | Pi% |
| 11-1 | –751057.5 | 0.0013 | 19.25 |
| 11-2 | –751057.29 | 0.2121 | 13.44 |
| 11-3 | –751057.11 | 0.3985 | 9.78 |
| 11-4 | –751056.95 | 0.5528 | 7.51 |
| 11-5 | –751057.5 | 0 | 19.29 |
| 11-6 | –751057.29 | 0.2127 | 13.42 |
| 11-7 | –751057.11 | 0.3966 | 9.81 |
| 11-8 | –751056.95 | 0.5541 | 7.5 |
| a which related to the most stable conformer. b Boltzmann weighting factor (Pi%) based on △G. |
|||
Conformers 11-1 (19.3%), 11-2 (13.4%), 11-3 (9.8%), 11-4 (7.5%), 11-5 (19.3%), 11-6 (13.4%), 11-7 (9.8%) and 11-8 (7.5%) are significantly populated at room temperature. The differences among the eight conformers resulted from the orientation of the isopropyl, sulfur atom and hydroxyl group. The ground-state energies between 11-1 and 11-5, 11-2 and 11-6, 11-3 and 11-7, or 11-4 and 11-8 are similar and only a slight difference between the orientations of the sulfur atom in a six-membered heterocyclic group was observed. The sulfur atoms of the first row conformations are below the plane while those of the second line conformations are above the plane.
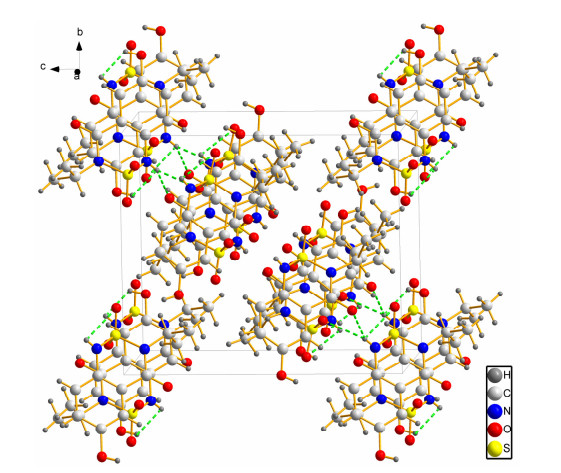
For a deeper insight into the structure characteristics of the designed analogs, X-ray structure analysis for 11 was performed. Crystals of 11 were grown by slow evaporation of N, N-dimethylformamide under ambient conditions, and suitable crystals were obtained for crystallographic analysis. The measured values revealed that 11 crystallizes in a triclinic system, space group P
DownLoad:
CSV
| D–H…A | D–H | H…A | D…A | D–H…A |
| O(3)–H(1O)…O(11)ⅰ | 0.83(2) | 1.97(2) | 2.785(3) | 170(3) |
| N(5)–H(2N)…O(7)ⅱ | 0.74(4) | 2.18(4) | 2.897(3) | 165(4) |
| O(8)–H(2O)…O(3)ⅲ | 0.82(3) | 2.14(3) | 2.897(3) | 154(4) |
| N(2)–H(3N)…O(6)ⅳ | 0.70(4) | 2.49(4) | 3.06(3) | 140(4) |
| N(2)–H(3N…O(11)ⅴ | 0.70(4) | 2.49(4) | 3.05(3) | 139(4) |
| O(12)–H(3O)…O(4)ⅵ | 0.79(3) | 2.39(3) | 3.093(3) | 150(4) |
| Symmetry code: (ⅰ) x, 1+y, z; (ⅱ) 1–x, 2–y, 1–z; (ⅲ) x, –1+y, 1+z; (ⅳ) 1+x, y, –1+z; (ⅴ) 1–x, 1–y, –z; (ⅵ) x, –1+y, z | ||||
The crystal structure of 11 was compared with the DFT-optimized structures. Among all the conformers, 11-1, 11-6 and 11-8 are in accordance with molecules a, b and c, respectively. Some selected experimental and calculated geometry parameters for 11 are selected, as listed in Tables 3~5.
DownLoad:
CSV
| Bond distances (Å) | Exp.a | Calcd.b | Difference |
| C(1)–N(2) | 1.4 | 1.41 | 0.01 |
| C(4)–O(3) | 1.4 | 1.36 | –0.04 |
| C(7)–O(4) | 1.21 | 1.22 | 0.01 |
| C(7)–N(1) | 1.39 | 1.4 | 0.01 |
| C(8)–N(1) | 1.5 | 1.51 | 0.01 |
| O(1)–S(1) | 1.41 | 1.46 | 0.05 |
| O(2)–S(1) | 1.41 | 1.45 | 0.04 |
| N(2)–S(1) | 1.67 | 1.67 | 0 |
| N(1)–S(1) | 1.6 | 1.71 | 0.11 |
| Bond angle (°) | Exp.a | Calcd.b | Difference |
| O(4)–C(7)–N(1) | 120.22 | 120.37 | 0.15 |
| C(7)–N(1)–S(1) | 120.75 | 120.68 | –0.07 |
| C(8)–N(1)–S(1) | 114.09 | 115.94 | 1.85 |
| C(1)–N(2)–S(1) | 119.32 | 116.79 | –2.53 |
| O(1)–S(1)–O(2) | 117.99 | 118.78 | 0.79 |
| O(1)–S(1)–N(2) | 111.48 | 112.36 | 0.88 |
| O(2)–S(1)–N(2) | 107.92 | 107.37 | –0.55 |
| O(1)–S(1)–N(1) | 108 | 109.34 | 1.34 |
| N(2)–S(1)–N(1) | 100.7 | 97.53 | –3.17 |
| O(2)–S(1)–N(1) | 109.35 | 109.36 | 0.01 |
| Torsion angles (Å) | Exp.a | Calcd.b | Difference |
| C(2)–C(1)–C(6)–C(5) | 1.94 | –0.53 | –2.47 |
| C(1)–C(6)–C(7)–N(1) | 6.64 | 10.74 | 4.1 |
| O(4)–C(7)–N(1)–C(8) | –5.24 | –0.81 | 4.43 |
| O(4)–C(7)–N(1)–S(1) | –163.62 | –164.73 | –1.11 |
| C(6)–C(1)–N(2)–S(1) | –33.43 | –38.3 | –4.87 |
| C(9)–C(8)–N(1)–S(1) | –135.83 | –130.4 | 5.43 |
| C(6)–C(7)–N(1)–S(1) | 17.73 | 17.41 | –0.32 |
| C(10)–C(8)–N(1)–S(1) | 91.63 | 101.38 | 9.75 |
| C(1)–N(2)–S(1)–O(1) | –65.52 | –59.38 | 6.14 |
| C(1)–N(2)–S(1)–O(2) | 163.42 | 168.23 | 4.81 |
| C(1)–N(2)–S(1)–N(1) | 48.82 | 55.17 | 6.35 |
| C(7)–N(1)–S(1)–O(1) | 75.72 | 71.69 | –4.03 |
| C(8)–N(1)–S(1)–O(1) | –84.42 | –93.08 | –8.66 |
| C(7)–N(1)–S(1)–O(2) | –154.72 | –156.69 | –1.97 |
| C(8)–N(1)–S(1)–O(2) | 45.22 | 38.53 | –6.69 |
| C(7)–N(1)–S(1)–N(2) | –41.32 | –45.24 | –3.92 |
| C(8)–N(1)–S(1)–N(2) | 158.67 | 149.98 | –8.69 |
| a: Experimental geometry parameters for molecule a; b: Calculated geometry parameters for conformer 11-1 | |||
DownLoad:
CSV
| Bond distances (Å) | Exp.a | Calcd.b | Difference |
| C(11)–N(3) | 1.4 | 1.41 | 0.01 |
| C(14)–O(8) | 1.36 | 1.36 | 0 |
| C(17)–O(7) | 1.21 | 1.22 | 0.01 |
| C(17)–N(4) | 1.38 | 1.41 | 0.03 |
| C(18)–N(4) | 1.5 | 1.51 | 0.01 |
| N(3)–S(2) | 1.61 | 1.68 | 0.07 |
| O(6)–S(2) | 1.41 | 1.46 | 0.05 |
| O(5)–S(2) | 1.4 | 1.45 | 0.05 |
| N(4)–S(2) | 1.67 | 1.7 | 0.03 |
| Bond angle (°) | Exp.a | Calcd.b | Difference |
| C(11)–N(3)–S(2) | 117.522 | 116.51 | –1.012 |
| C(17)–N(4)–S(2) | 120.97 | 120.43 | –0.54 |
| C(18)–N(4)–S(2) | 120.02 | 119.21 | –0.81 |
| O(5)–S(2)–O(6) | 118.35 | 118.94 | 0.59 |
| O(5)–S(2)–N(3) | 108.97 | 107.26 | –1.71 |
| O(6)–S(2)–N(3) | 110.94 | 112.03 | 1.09 |
| O(5)–S(2)–N(4) | 108.7 | 109.86 | 1.16 |
| O(6)–S(2)–N(4) | 108.09 | 109.11 | 1.02 |
| O(7)–C(17)–N(4) | 121.23 | 120.49 | –0.74 |
| N(3)–S(2)–N(4) | 100.23 | 97.53 | –2.7 |
| Torsion angles (Å) | Exp.a | Calcd.b | Difference |
| C(15)–C(16)–C(11)–C(12) | –1.34 | 0.37 | 1.71 |
| C(11)–C(16)–C(17)–N(4) | –11.14 | –11.83 | –0.69 |
| C(16)–C(11)–N(3)–S(2) | 37.73 | 38.71 | 0.98 |
| O(7)–C(17)–N(4)–C(18) | 3.54 | –1.51 | –5.05 |
| O(7)–C(17)–N(4)–S(2) | 168.62 | 165.5 | –3.12 |
| C(16)–C(17)–N(4)–S(2) | –12.83 | –16.75 | –3.92 |
| C(19)–C(18)–N(4)–S(2) | –41.34 | –50.05 | –8.71 |
| C(20)–C(18)–N(4)–S(2) | 90.83 | 79.12 | –11.71 |
| C(11)–N(3)–S(2)–O(5) | –166.12 | –169.61 | –3.49 |
| C(11)–N(3)–S(2)–O(6) | 61.92 | 58.14 | –3.78 |
| C(11)–N(3)–S(2)–N(4) | –52.22 | –56.04 | –3.82 |
| C(17)–N(4)–S(2)–O(5) | 154.42 | 156.89 | 2.47 |
| C(18)–N(4)–S(2)–O(5) | –40.83 | –36.1 | 4.73 |
| C(17)–N(4)–S(2)–O(6) | –76.02 | –71.07 | 4.95 |
| C(18)–N(4)–S(2)–O(6) | 88.92 | 95.94 | 7.02 |
| C(17)–N(4)–S(2)–N(3) | 40.22 | 45.43 | 5.21 |
| C(18)–N(4)–S(2)–N(3) | –155.02 | –147.56 | 7.46 |
| a: Experimental geometry parameters for molecule b; b: Calculated geometry parameters for conformer 11-6. | |||
DownLoad:
CSV
| Bond distances (Å) | Exp.a | Calcd.b | Difference |
| N(5)–S(3) | 1.6 | 1.68 | 0.08 |
| C(24)–O(12) | 1.36 | 1.36 | 0 |
| C(27)–O(11) | 1.23 | 1.22 | –0.01 |
| C(27)–N(6) | 1.37 | 1.41 | 0.04 |
| C(28)–N(6) | 1.5 | 1.51 | 0.01 |
| C(21)–N(5) | 1.4 | 1.41 | 0.01 |
| N(6)–S(3) | 1.68 | 1.7 | 0.02 |
| O(10)–S(3) | 1.41 | 1.46 | 0.05 |
| O(9)–S(3) | 1.41 | 1.45 | 0.04 |
| Bond angle (°) | Exp.a | Calcd.b | Difference |
| C(21)–N(5)–S(3) | 118.52 | 116.37 | –2.15 |
| O(11)–C(27)–N(6) | 120.82 | 120.46 | –0.36 |
| N(5)–S(3)–N(6) | 100.13 | 97.51 | –2.62 |
| C(28)–N(6)–S(3) | 119.15 | 119.12 | –0.03 |
| O(10)–S(3)–O(9) | 118.99 | 118.93 | –0.06 |
| O(10)–S(3)–N(5) | 111.26 | 112.04 | 0.78 |
| O(9)–S(3)–N(5) | 108.81 | 107.24 | –1.57 |
| O(10)–S(3)–N(6) | 107.59 | 109.14 | 1.55 |
| O(9)–S(3)–N(6) | 108.34 | 109.86 | 1.52 |
| C(27)–N(6)–S(3) | 120.59 | 120.51 | –0.08 |
| Torsion angles (Å) | Exp.a | Calcd.b | Difference |
| C(25)–C(26)–C(21–C(22) | –0.64 | 0.42 | 1.06 |
| C(21)–C(26)–C(27)–N(6) | –14.04 | –12.34 | 1.7 |
| C(26)–C(21)–N(5)–S(3) | 35.63 | 39.24 | 3.61 |
| O(11)–C(27)–N(6)–C(28) | 3.44 | –1.17 | –4.61 |
| O(11)–C(27)–N(6)–S(3) | 169.55 | 166.07 | –3.48 |
| C(26)–C(27)–N(6)–S(3) | –11.33 | –16.1 | –4.77 |
| C(30)–C(28)–N(6)–S(3) | 84.13 | 79.05 | –5.08 |
| C(29)–C(28)–N(6)–S(3) | –47.73 | –50.13 | –2.4 |
| C(21)–N(5)–S(3)–O(10) | 61.92 | 57.81 | –4.11 |
| C(21)–N(5)–S(3)–O(9) | –165.12 | –169.97 | –4.85 |
| C(21)–N(5)–S(3)–N(6) | –51.62 | –56.41 | –4.79 |
| C27)–N(6)–S(3)–O(10) | –76.72 | –71.31 | 5.41 |
| C(28)–N(6)–S(3)–O(10) | 89.42 | 95.93 | 6.51 |
| C(27)–N(6)–S(3)–O(9) | 153.42 | 156.64 | 3.22 |
| C(28)–N(6)–S(3)–O(9) | –40.52 | –36.12 | 4.4 |
| C(27)–N(6)–S(3)–N(5) | 39.62 | 45.21 | 5.59 |
| C(28)–N(6)–S(3)–N(5) | –154.32 | –147.55 | 6.77 |
| a: Experimental geometry parameters for molecule c; b: Calculated geometry parameters for conformer 11-8 | |||
Compound 11 crystallizes in space group P1 with Z = 6, and the crystal structure of conformer a is shown in Fig. S2. In conformer a, the bond lengths of S(1)=O(1) (1.46 Å) and S(1)=O(2) (1.45 Å) are close to the normal S=O bond (1.43 Å). The bond lengths of N(1)–S(1) (1.71 Å) and N(2)–S(1) (1.67 Å) are longer than the normal single N–S bond (1.60 Å). The bond length of C(7)=O(4) (1.22 Å) is close to the normal C=O bond (1.20 Å) and shorter than C(4)–O(3) (1.36 Å). The O(1)–S(1)–N(2), O(2)–S(1)–N(2), O(1)–S(1)–N(1) and O(2)–S(1)–N(1) bond angles are 112.36º, 107.37º, 109.34º and 109.36º, respectively. The torsion angles of C(2)–C(1)–C(6)–C(5), C(1)–C(6)–C(7)–N(1) and C(6)–C(1)–N(2)–S(1) are –0.53º, 10.74º and –38.3º, respectively, which indicate the phenyl and thiadiazine rings do not share a common plane.
Conformer b has similar spatial structure to a, and its crystal structure is shown in Fig. S2. In conformer b, the bond lengths of S(2)=O(5) (1.45 Å) and S(2)=O(6) (1.46 Å) are close to that of the normal single S=O bond (1.43 Å). The bond lengths of N(4)–S(2) (1.70 Å) and N(3)–S(2) (1.68 Å) are longer than the normal single N–S bond (1.60 Å). The C(17)=O(7) bond in 1.22 Å is close to normal single C=O bond (1.20 Å) and shorter than C(14)–O(8) (1.36 Å). The O(5)–S(2)–N(3), O(6)–S(2)–N(3), O(5)–S(2)–N(4) and O(6)–S(2)–N(4) bond angles are 107.26º, 112.03º, 109.86º and 109.11º, respectively. The torsion angles of C(15)–C(16)–C(11)–C(12), C(11)–C(16)–C(17)–N(4) and C(16)–C(11)–N(3)– S(2) are 0.37º, –11.83º and 38.71º correspondingly, which indicate the phenyl and thiadiazine rings do not share a common plane.
For conformer c, the spatial structure is similar to conformer of a and b. The crystal structure of conformer c is shown in Fig. S2. In c, the bond lengths of S(3)=O(9) (1.45 Å) and S(2)=O(10) (1.46 Å) are close to that of the normal single S=O bond (1.43 Å). The bonds of N(6)–S(3) (1.70 Å) and N(5)–S(3) (1.68 Å) are longer than the normal single N–S bond (1.60 Å). The C(27)=O(11) bond in 1.22 Å is close to the normal single C=O bond (1.20 Å) and shorter than C(24)–O(12) (1.36 Å). The O(10)–S(3)–N(5), O(9)–S(3)–N(5), O(10)–S(3)–N(6) and O(9)–S(3)– N(6) bond angles are 112.04º, 107.24º, 109.14º and 109.86º, respectively. The torsion angles of C(25)–C(26)–C(21–C(22), C(21)–C(26)–C(27)–N(6) and C(26)–C(21)–N(5)–S(3) are 0.42º, –12.34º and 39.24º, respectively, which indicate the phenyl and thiadiazine rings do not share a common plane.
In order to get the information about the region from which conformers 11-1 (the major conformer) can show intermolecular interactions, the MEP was investigated by the B3LYP/6311G(d, p) method. In the MEP map, different electrostatic potentials at the surface are represented by different colors, and the potential increases in the order of red < orange < yellow < green < blue. As shown in Fig. S3, the O4 atom in the carbonyl group and the O(1)=S(1)=O(2) group of conformer 11-1 were surrounded by negative charge, which indicated the presence of some sites for nucleophilic attack. In addition, the positive regions were localized on the H atoms in the hydroxyl group and the sulfonamide group. Considering the calculated results, the MEP map revealed that the negative potential sites were on the electronegative, and the positive sites around the N–H bands, which confirmed the existence of intermolecular N–H···O=C interactions (intermolecular hydrogen bonding) that made the structure more stable in the solid state.
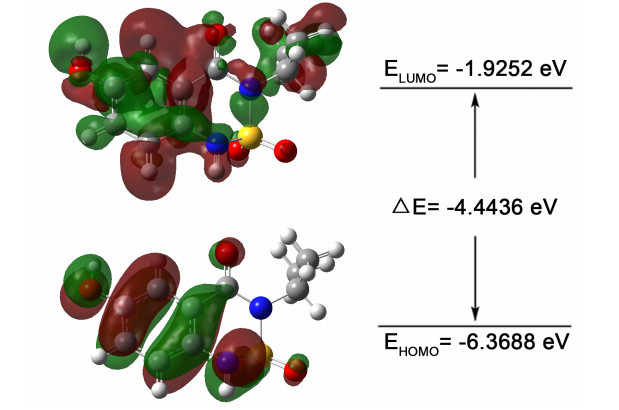
To investigate the chemical stability of conformer 11-1, the energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) together with their orbital energy gap were calculated using the B3LYP/6-311G(d, p) method. Fig. 3 shows a pictorial illustration of FMOs and the respective positive and negative regions represented in red and green. The large HOMO-LUMO gap automatically implies the high excitation energies of the excited states, good stability and large chemical hardness for the calculated conformer.
This paper reports the synthesis of 6-hydroxybentazon, along with its crystal structure determined by single-crystal X-ray diffraction analysis. Moreover, the optimized structures of the molecule were calculated by DFT using the B3LYP method with the 6-311G(d, p) basis set, and compared with the X-ray structures.
Boivin, A.; Cherrier, R.; Perrin-Ganier, C.; Schiavon, M. Time effect on bentazone sorption and degradation in soil. Pest. Manag. Sci. 2004, 60, 809–814. doi: 10.1002/ps.889
Chiarandini, J. P.; Escandar, G. M. Nylon membrane as a fluorimetric probe for the herbicide bentazone. Anal. Bioanal. Chem. 2012, 402, 2221–2225. doi: 10.1007/s00216-011-5682-z
Bruzzoniti, M. C.; De Carlo, R. M.; Rivoira, L.; Del Bubba, M.; Pavan, M.; Riatti, M.; Onida, B. Adsorption of bentazone herbicide onto mesoporous silica: application to environmental water purification. Environ. Sci. Pollut. Res. 2016, 23, 5399–5409. doi: 10.1007/s11356-015-5755-1
Odjegba, V. J.; Adeniran, R. A. Bentazone herbicide induces genotoxic effect and physiological disorders in non-targeted Allium cepa L. Indian. J. Plant. Physiol. 2015, 20, 375–379. doi: 10.1007/s40502-015-0168-1
Martinez, A.; Castro, A.; Gil, C.; Miralpeix, M.; Segarra, V.; Doménech, T.; Beleta, J.; Palacios, J. M.; Ryder, H.; Miró, X.; Bonet, C.; Casacuberta, J. M.; Azorín, F.; Puigdoménech, P. Benzyl derivatives of 2, 1, 3-benzo-and benzothieno [3, 2-a] thiadiazine 2, 2-dioxides: first phosphodiesterase 7 inhibitors. J. Med. Chem. 2000, 43, 683–689. doi: 10.1021/jm990382n
Castro, A.; Abasolo, M. I.; Gil, C.; Segarra, V.; Martinez, A. CoMFA of benzyl derivatives of 2, 1, 3-benzo and benzothieno [3, 2-a] thiadiazine 2, 2-dioxides: clues for the design of phosphodiesterase 7 inhibitors. Eur. J. Med. Chem. 2001, 36, 333–338. doi: 10.1016/S0223-5234(01)01227-2
Qi, X.; Fu, Z. T.; Li, L.; Hu, X. M.; Sun, H. B. A green synthesis technique of bentazone without discharging waste water Vols. 396-398. Tokyo: Advanced Materials Research, 2012, 2023–2026.
Le, Y.; Zhao, C. S.; Zhou, Z. X.; Liu, L.; Huang, Z. Y.; Teng, M. G. Method for preparing 6-hydroxybentazone. CN patent, C07. 106478548. 2017-05-08.
Sheldrick, G. M. SHELXS-97, Program for the Solution of Crystal Structures. University of Göttingen, Germany 1997.
Mori, T.; Inoue, Y.; Grimme, S. Time dependent density functional theory calculations for electronic circular dichroism spectra and optical rotations of conformationally flexible chiral donor-acceptor dyad. J. Org. Chem. 2006, 71, 9797–9806. doi: 10.1021/jo061855i
Marchesan, D.; Coriani, S.; Forzato, C.; Nitti, P.; Pitacco, G.; Ruud, K. Optical rotation calculation of a highly flexible molecule: the case of paraconic acid. J. Phys. Chem. A 2005, 109, 1449–1453. doi: 10.1021/jp047108b
Kwit, M.; Sharma, N. D.; Boyd, D. R.; Gawronski, J. Determination of absolute configuration of conformationally flexible cis-dihydrodiol metabolites: effect of diene substitution pattern on the circular dichroism spectra and optical rotations. Chirality 2008, 20, 609–620. doi: 10.1002/chir.20471
Deppmeier, B. J.; Driessen, A. J.; Hehre, T. S.; Hehre, W. J.; Johnson, J. A.; Klunzinger, P. E.; Kong, J. Wavefunction Inc, Spartan'08., Irvine, CA 2008.
Bosnich, B. Molecular mechanics force fields for cyclopentadienyl complexes. Chem. Soc. Rev. 1994, 23, 387–395. doi: 10.1039/cs9942300387
Huang, N.; Kalyanaraman, C.; Bernacki, K.; Jacobson, M. P. Molecular mechanics methods for predicting protein-ligand binding. Phys. Chem. Chem. Phys. 2006, 8, 5166–5177. doi: 10.1039/B608269F
Koch, W.; Holthausen, M. C.; Holthausen, M. C. BUCHER-A chemist's guide to density functional theory. Angew. Chem. Ger. Edit. 2001, 113, 989–989.
Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem Rev. 2003, 103, 1793–1874. doi: 10.1021/cr990029p
Saberikia, I.; Safaei, E.; Kowsari, M. H.; Lee, Y. I.; Cotic, P.; Bruno, G.; Rudbari, H. A. A new iron(Ⅲ) complex of glycine derivative of amine-chloro substituted phenol ligand: synthesis, characterization and catechol dioxygenase activity. J. Mol. Struct. 2012, 1029, 60–67. doi: 10.1016/j.molstruc.2012.06.047

Figure 3 The highest occupied and lowest unoccupied molecular orbitals of conformer 11-1 obtained by the DFT/6311G (d, p) method
Table 1. Gibbs Free Energies (G), Relative Gibbs Free Energies (△G)a and Boltzmann Weighting Factor (P%)b of 11 Conformers by Using the DFT/B3LYP/6-311G(d, p) Method
| Conformer | G(kcal mol-1) | △G(kcal mol-1) | Pi% |
| 11-1 | –751057.5 | 0.0013 | 19.25 |
| 11-2 | –751057.29 | 0.2121 | 13.44 |
| 11-3 | –751057.11 | 0.3985 | 9.78 |
| 11-4 | –751056.95 | 0.5528 | 7.51 |
| 11-5 | –751057.5 | 0 | 19.29 |
| 11-6 | –751057.29 | 0.2127 | 13.42 |
| 11-7 | –751057.11 | 0.3966 | 9.81 |
| 11-8 | –751056.95 | 0.5541 | 7.5 |
| a which related to the most stable conformer. b Boltzmann weighting factor (Pi%) based on △G. |
|||
 下载: 导出CSV
下载: 导出CSV
Table 2. Hydrogen-bond Geometry (Å, °) of 11
| D–H…A | D–H | H…A | D…A | D–H…A |
| O(3)–H(1O)…O(11)ⅰ | 0.83(2) | 1.97(2) | 2.785(3) | 170(3) |
| N(5)–H(2N)…O(7)ⅱ | 0.74(4) | 2.18(4) | 2.897(3) | 165(4) |
| O(8)–H(2O)…O(3)ⅲ | 0.82(3) | 2.14(3) | 2.897(3) | 154(4) |
| N(2)–H(3N)…O(6)ⅳ | 0.70(4) | 2.49(4) | 3.06(3) | 140(4) |
| N(2)–H(3N…O(11)ⅴ | 0.70(4) | 2.49(4) | 3.05(3) | 139(4) |
| O(12)–H(3O)…O(4)ⅵ | 0.79(3) | 2.39(3) | 3.093(3) | 150(4) |
| Symmetry code: (ⅰ) x, 1+y, z; (ⅱ) 1–x, 2–y, 1–z; (ⅲ) x, –1+y, 1+z; (ⅳ) 1+x, y, –1+z; (ⅴ) 1–x, 1–y, –z; (ⅵ) x, –1+y, z | ||||
下载: 导出CSV
Table 3. Selected Bond Lengths, Angles and Torsion Angles for Molecule a and Conformation 11-1
| Bond distances (Å) | Exp.a | Calcd.b | Difference |
| C(1)–N(2) | 1.4 | 1.41 | 0.01 |
| C(4)–O(3) | 1.4 | 1.36 | –0.04 |
| C(7)–O(4) | 1.21 | 1.22 | 0.01 |
| C(7)–N(1) | 1.39 | 1.4 | 0.01 |
| C(8)–N(1) | 1.5 | 1.51 | 0.01 |
| O(1)–S(1) | 1.41 | 1.46 | 0.05 |
| O(2)–S(1) | 1.41 | 1.45 | 0.04 |
| N(2)–S(1) | 1.67 | 1.67 | 0 |
| N(1)–S(1) | 1.6 | 1.71 | 0.11 |
| Bond angle (°) | Exp.a | Calcd.b | Difference |
| O(4)–C(7)–N(1) | 120.22 | 120.37 | 0.15 |
| C(7)–N(1)–S(1) | 120.75 | 120.68 | –0.07 |
| C(8)–N(1)–S(1) | 114.09 | 115.94 | 1.85 |
| C(1)–N(2)–S(1) | 119.32 | 116.79 | –2.53 |
| O(1)–S(1)–O(2) | 117.99 | 118.78 | 0.79 |
| O(1)–S(1)–N(2) | 111.48 | 112.36 | 0.88 |
| O(2)–S(1)–N(2) | 107.92 | 107.37 | –0.55 |
| O(1)–S(1)–N(1) | 108 | 109.34 | 1.34 |
| N(2)–S(1)–N(1) | 100.7 | 97.53 | –3.17 |
| O(2)–S(1)–N(1) | 109.35 | 109.36 | 0.01 |
| Torsion angles (Å) | Exp.a | Calcd.b | Difference |
| C(2)–C(1)–C(6)–C(5) | 1.94 | –0.53 | –2.47 |
| C(1)–C(6)–C(7)–N(1) | 6.64 | 10.74 | 4.1 |
| O(4)–C(7)–N(1)–C(8) | –5.24 | –0.81 | 4.43 |
| O(4)–C(7)–N(1)–S(1) | –163.62 | –164.73 | –1.11 |
| C(6)–C(1)–N(2)–S(1) | –33.43 | –38.3 | –4.87 |
| C(9)–C(8)–N(1)–S(1) | –135.83 | –130.4 | 5.43 |
| C(6)–C(7)–N(1)–S(1) | 17.73 | 17.41 | –0.32 |
| C(10)–C(8)–N(1)–S(1) | 91.63 | 101.38 | 9.75 |
| C(1)–N(2)–S(1)–O(1) | –65.52 | –59.38 | 6.14 |
| C(1)–N(2)–S(1)–O(2) | 163.42 | 168.23 | 4.81 |
| C(1)–N(2)–S(1)–N(1) | 48.82 | 55.17 | 6.35 |
| C(7)–N(1)–S(1)–O(1) | 75.72 | 71.69 | –4.03 |
| C(8)–N(1)–S(1)–O(1) | –84.42 | –93.08 | –8.66 |
| C(7)–N(1)–S(1)–O(2) | –154.72 | –156.69 | –1.97 |
| C(8)–N(1)–S(1)–O(2) | 45.22 | 38.53 | –6.69 |
| C(7)–N(1)–S(1)–N(2) | –41.32 | –45.24 | –3.92 |
| C(8)–N(1)–S(1)–N(2) | 158.67 | 149.98 | –8.69 |
| a: Experimental geometry parameters for molecule a; b: Calculated geometry parameters for conformer 11-1 | |||
下载: 导出CSV
Table 4. Selected Bond Lengths, Angles and Torsion Angles for Molecule b and Conformation 11-6
| Bond distances (Å) | Exp.a | Calcd.b | Difference |
| C(11)–N(3) | 1.4 | 1.41 | 0.01 |
| C(14)–O(8) | 1.36 | 1.36 | 0 |
| C(17)–O(7) | 1.21 | 1.22 | 0.01 |
| C(17)–N(4) | 1.38 | 1.41 | 0.03 |
| C(18)–N(4) | 1.5 | 1.51 | 0.01 |
| N(3)–S(2) | 1.61 | 1.68 | 0.07 |
| O(6)–S(2) | 1.41 | 1.46 | 0.05 |
| O(5)–S(2) | 1.4 | 1.45 | 0.05 |
| N(4)–S(2) | 1.67 | 1.7 | 0.03 |
| Bond angle (°) | Exp.a | Calcd.b | Difference |
| C(11)–N(3)–S(2) | 117.522 | 116.51 | –1.012 |
| C(17)–N(4)–S(2) | 120.97 | 120.43 | –0.54 |
| C(18)–N(4)–S(2) | 120.02 | 119.21 | –0.81 |
| O(5)–S(2)–O(6) | 118.35 | 118.94 | 0.59 |
| O(5)–S(2)–N(3) | 108.97 | 107.26 | –1.71 |
| O(6)–S(2)–N(3) | 110.94 | 112.03 | 1.09 |
| O(5)–S(2)–N(4) | 108.7 | 109.86 | 1.16 |
| O(6)–S(2)–N(4) | 108.09 | 109.11 | 1.02 |
| O(7)–C(17)–N(4) | 121.23 | 120.49 | –0.74 |
| N(3)–S(2)–N(4) | 100.23 | 97.53 | –2.7 |
| Torsion angles (Å) | Exp.a | Calcd.b | Difference |
| C(15)–C(16)–C(11)–C(12) | –1.34 | 0.37 | 1.71 |
| C(11)–C(16)–C(17)–N(4) | –11.14 | –11.83 | –0.69 |
| C(16)–C(11)–N(3)–S(2) | 37.73 | 38.71 | 0.98 |
| O(7)–C(17)–N(4)–C(18) | 3.54 | –1.51 | –5.05 |
| O(7)–C(17)–N(4)–S(2) | 168.62 | 165.5 | –3.12 |
| C(16)–C(17)–N(4)–S(2) | –12.83 | –16.75 | –3.92 |
| C(19)–C(18)–N(4)–S(2) | –41.34 | –50.05 | –8.71 |
| C(20)–C(18)–N(4)–S(2) | 90.83 | 79.12 | –11.71 |
| C(11)–N(3)–S(2)–O(5) | –166.12 | –169.61 | –3.49 |
| C(11)–N(3)–S(2)–O(6) | 61.92 | 58.14 | –3.78 |
| C(11)–N(3)–S(2)–N(4) | –52.22 | –56.04 | –3.82 |
| C(17)–N(4)–S(2)–O(5) | 154.42 | 156.89 | 2.47 |
| C(18)–N(4)–S(2)–O(5) | –40.83 | –36.1 | 4.73 |
| C(17)–N(4)–S(2)–O(6) | –76.02 | –71.07 | 4.95 |
| C(18)–N(4)–S(2)–O(6) | 88.92 | 95.94 | 7.02 |
| C(17)–N(4)–S(2)–N(3) | 40.22 | 45.43 | 5.21 |
| C(18)–N(4)–S(2)–N(3) | –155.02 | –147.56 | 7.46 |
| a: Experimental geometry parameters for molecule b; b: Calculated geometry parameters for conformer 11-6. | |||
下载: 导出CSV
Table 5. Selected Bond Lengths, Angles and Torsion Angles for Molecule c and Conformation 11-8
| Bond distances (Å) | Exp.a | Calcd.b | Difference |
| N(5)–S(3) | 1.6 | 1.68 | 0.08 |
| C(24)–O(12) | 1.36 | 1.36 | 0 |
| C(27)–O(11) | 1.23 | 1.22 | –0.01 |
| C(27)–N(6) | 1.37 | 1.41 | 0.04 |
| C(28)–N(6) | 1.5 | 1.51 | 0.01 |
| C(21)–N(5) | 1.4 | 1.41 | 0.01 |
| N(6)–S(3) | 1.68 | 1.7 | 0.02 |
| O(10)–S(3) | 1.41 | 1.46 | 0.05 |
| O(9)–S(3) | 1.41 | 1.45 | 0.04 |
| Bond angle (°) | Exp.a | Calcd.b | Difference |
| C(21)–N(5)–S(3) | 118.52 | 116.37 | –2.15 |
| O(11)–C(27)–N(6) | 120.82 | 120.46 | –0.36 |
| N(5)–S(3)–N(6) | 100.13 | 97.51 | –2.62 |
| C(28)–N(6)–S(3) | 119.15 | 119.12 | –0.03 |
| O(10)–S(3)–O(9) | 118.99 | 118.93 | –0.06 |
| O(10)–S(3)–N(5) | 111.26 | 112.04 | 0.78 |
| O(9)–S(3)–N(5) | 108.81 | 107.24 | –1.57 |
| O(10)–S(3)–N(6) | 107.59 | 109.14 | 1.55 |
| O(9)–S(3)–N(6) | 108.34 | 109.86 | 1.52 |
| C(27)–N(6)–S(3) | 120.59 | 120.51 | –0.08 |
| Torsion angles (Å) | Exp.a | Calcd.b | Difference |
| C(25)–C(26)–C(21–C(22) | –0.64 | 0.42 | 1.06 |
| C(21)–C(26)–C(27)–N(6) | –14.04 | –12.34 | 1.7 |
| C(26)–C(21)–N(5)–S(3) | 35.63 | 39.24 | 3.61 |
| O(11)–C(27)–N(6)–C(28) | 3.44 | –1.17 | –4.61 |
| O(11)–C(27)–N(6)–S(3) | 169.55 | 166.07 | –3.48 |
| C(26)–C(27)–N(6)–S(3) | –11.33 | –16.1 | –4.77 |
| C(30)–C(28)–N(6)–S(3) | 84.13 | 79.05 | –5.08 |
| C(29)–C(28)–N(6)–S(3) | –47.73 | –50.13 | –2.4 |
| C(21)–N(5)–S(3)–O(10) | 61.92 | 57.81 | –4.11 |
| C(21)–N(5)–S(3)–O(9) | –165.12 | –169.97 | –4.85 |
| C(21)–N(5)–S(3)–N(6) | –51.62 | –56.41 | –4.79 |
| C27)–N(6)–S(3)–O(10) | –76.72 | –71.31 | 5.41 |
| C(28)–N(6)–S(3)–O(10) | 89.42 | 95.93 | 6.51 |
| C(27)–N(6)–S(3)–O(9) | 153.42 | 156.64 | 3.22 |
| C(28)–N(6)–S(3)–O(9) | –40.52 | –36.12 | 4.4 |
| C(27)–N(6)–S(3)–N(5) | 39.62 | 45.21 | 5.59 |
| C(28)–N(6)–S(3)–N(5) | –154.32 | –147.55 | 6.77 |
| a: Experimental geometry parameters for molecule c; b: Calculated geometry parameters for conformer 11-8 | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


