Chemistry and Environment Science College, Inner Mongolia Normal University, Inner Mongolia Key Laboratory of Green Catalysis, Hohhot, Inner Mongolia 010022, China
b.
School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
Received Date:
18 April 2019 Accepted Date:
12 September 2019 Available Online:
01 February 2020
Fund Project:
Abstract:
Theoretical calculations of Double Hanging Ring Molecule (DHRM) [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge; n = 3, 5, 6, 7, 8; m = +1, –1, 0, +1, +2) were performed via Gaussian 09 with the method of Density Functional Theory (DFT). Geometrical optimization, Potential Energy surface Scan (PES), Degree of Aromaticity (DOA) and Nucleus Independent Chemical Shift (NICS) were computed to study the optimal structures and aromaticity of DHRMs. Ring Stretching Vibration Raman Spectroscopy (RSVRSF) was predicted to seek the relation between RSVRSF and aromaticity of DHRMs. The results show optimal structures of DHRMs[(GnHn–1m)(GnHn–1m)] (n = 3, 5~8); DA = 90° is the stable structure when n = 3, 7, 8; while n = 5 corresponds to DA = 30°, n = 6 corresponds to DA = 50°; the correlation between DOA and NICS of DHRMs is quadratic; the value of RSVRSF of DHRM approximates to its corresponding single ring molecule, which could act as characteristic frequency of ring molecule to identify its aromaticity; the correlation between RSVRSF and DOA is quadratic, and that between RSVRSF and NICS is linear.
Although extensive researches were performed on the structures and aromaticity of single ring aromatic molecules, such as GnHnm (G = C, Si, Ge, n = 3, 5, 6, 7, 8, m = +1, –1, 0, +1, +2), the molecules assembled by two or more single ring molecules were hardly found. Hanging Ring Molecules (HRM) consist of two or more rings bonded by G-G, and each of the ring remains its plane structure unchanged, and the number of π electrons of each ring conforms to Huckel 4n+2 rule[1].
Biphenyl is a vital HRM acting as an important organic raw material, which was widely used in the field of scientific research, pharmaceutical research, pesticide, dyestuff and liquid crystal material[2~5]. HRM are serial molecules that are analogous to biphenyl.
Until now, few literatures can be our reference about HRM, no matter in the aspect of experiment or theoretical prediction. In this article, we will introduce Double Hanging Ring Molecule (DHRM) [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5, 6, 7, 8, m = +1, –1, 0, +1, +2), which takes biphenyl as prototype and consists of its corresponding single ring molecules GnHnm (G = C, Si, Ge, n = 3, 5, 6, 7, 8, m = +1, –1, 0, +1, +2), to study its structure and the relation between its Raman spectroscopy and aromaticity.
In early days, our member Yanbo Guo[6] have studied a series of single ring molecules GnHnm (G = C, Si, Ge), and found there are high positive correlations between theoretical Nucleus Independent Chemical Shift (NICS) and the value of its Ring Stretching Vibration Raman Frequency (RSVRSF) of GnHnm. Recently, we have studied single ring molecules in the process of Bond Length Equalization, and Degree of Aromaticity (DOA) was proposed[2]. DOA has good correlation with NICS and RSVRSF among single ring molecules. These above conclusions are only concluded from single ring molecules. Whether is it suitable for Hanging Ring Molecules (HRM) or not? What is the relation between DOA and NICS of DHRMs? And what is the relation between RSVRSF and Aromaticity? All of them are deserved to be studied further.
To answer the questions mentioned above, we theoretically studied DHRM[(GnHn–1m)(GnHn–1m)] of their geometrical structures, electronic structures, magnetic properties and Aromaticities, using quantum chemistry package Gaussian 09 with the method of Density Functional Theory (B3LYP/6-311+g (d, p))[7]. We hope that the following results of DHRMs could be reference to the synthesis of series HRMs.
2.
CALCULATION METHODS
2.1
Geometrical optimization, NICS and Raman spectroscopy of DHRM
Geometrical optimization and Raman spectroscopy of Double Hanging Ring Molecule (DHRM) [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5, 6, 7, 8, m = +1, –1, 0, +1, +2) were done via Gaussian09 to get optimal structure and Raman spectroscopy[6] with the method of B3LYP/6-311+g (d, p)[2].
Based on the optimized structure of DHRM, we can set a series of Bq atoms at the center on one of the two rings and compute its NMR. The negative value of the NMR of Bq is the Nucleus Independent Chemical Shift (NICS) of the molecule, with the same method of B3LYP/6-311+g (d, p)[2] and the key word to be "NMR = GIAO".
With the optimal structure and Raman spectroscopy results, we can use GaussianView to observe the vibration mode of the molecule, and then determine the ring stretching frequency of the molecule, namely Ring Stretching Vibration Raman Frequency (RSVRSF)[6].
2.2
Calculation of Bond Length Equalization energy (EBLE), delocalized bond energy (EDBE) and Degree of Aromaticity (DOA)
Similar method would be applied to DHRM to calculate EBLE, EDBE and DOA as we have done on the single aromatic molecules before. The following is a brief introduction to these three quantities, and it should be focused on the results of DHRM. Calculation details could be referred from the published article "Bond Length Equalization with molecular Aromaticity — A new measurement of Aromaticity" of ours[8].
2.2.1
EBLE of DHRM
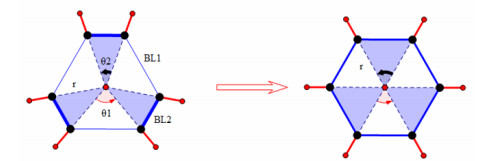
EBLE is the amount of energy that ring aromatic molecule decreased because of π electrons' delocalization; It is the difference between two single point energies of the same molecular formula with different structures. One is the structure in which all bond lengths of adjacent ring atoms are equal, and the other is that with unequal bond lengths[8], namely single and double bonds arranged alternately[2~3], just as Fig. 1 shows. On the basis of the optimized structure, by changing the radius angles, not only the single but also the double bond angles, we can get another molecular structure which corresponds to another single point energy. But how to change the radius angle is very important. We choose 0.8683 as the ratio of single and double bond lengths because it is the ratio of CC bonds of optimized C2H4 and C2H6[8]. On this ratio, the relationships between different lengths of ring atoms are obvious single and double bonds.
Figure 1
Figure 1.
An illustration of C6H6 in the process of BLE with its structure from left to right. Ring radius r, single bond radius angle $ {\mathit{\theta }}_{1} $, double bond radius angle $ {\mathit{\theta }}_{2} $, single bond length $ {\mathit{B}\mathit{L}}_{1} $, double bond length $ {\mathit{B}\mathit{L}}_{2} $
The calculation method of EBLE of DHRM is generalized from single molecules and applied to one of the two rings of DHRM.
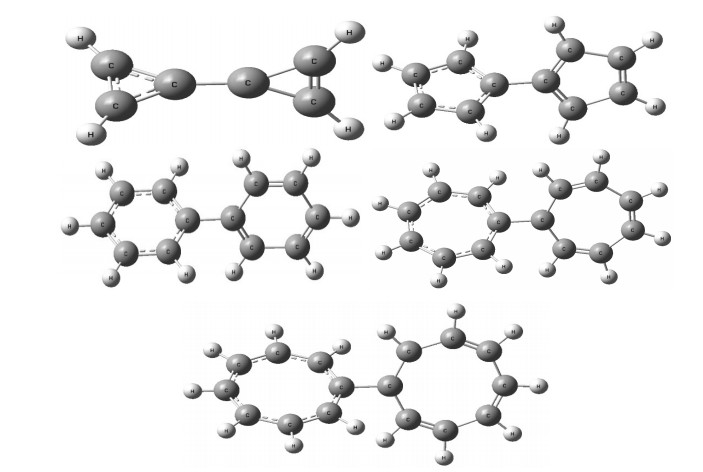
From Fig. 2 we can see that in each molecule every left ring has all equal bond lengths of adjacent rings, while the right one has interval equal bond lengths. And each structure of Fig. 4 is the molecule that has been optimized from Gaussian09, in which the adjacent bonds of the ring are equal, and two rings are symmetric with each other, so we choose one to change its bond length as it does in the single ring. EBLE of DHRM is the energy difference between the single point energyies of each molecule in Fig. 2 and that in Fig. 4. Each structure of Fig. 4 is the optimized molecule, based on which, by changing its radius angle of $ {\theta }_{1} $ and $ {\theta }_{2} $ to the ratio of single and double bond lengths equal to 0.8683, just as Fig. 1 illustrates, we can get the structure of Fig. 2 to calculate the EBLE of DHRM.
Figure 2
Figure 2.
An illustration of EBLE of DHRM [(CnHn-1m)(CnHn-1m)] (Right rings are single and double ring bonds alternatively, and left rings are all equal ring bonds. It should be compared with Fig. 4)
Figure 3a.
Splitting structure illustration of EDBE of [(C3H2+)(C3H2+)] (Violet atoms are the dummy atoms that are only helpful for comprehending, but make no difference for the results)
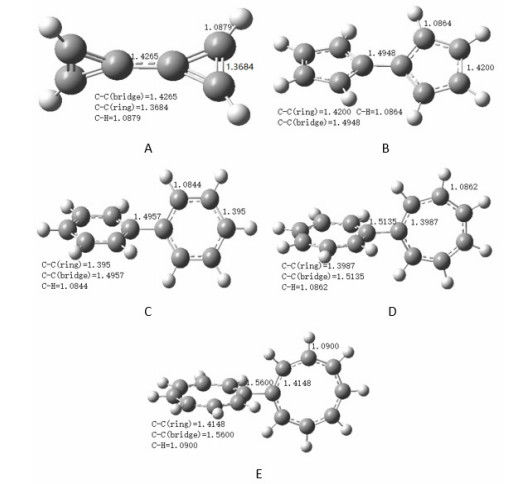
Figure 4.
Optimized structures of DHRM, A [(C3H2+)(C3H2+)], B [(C5H4-)(C5H4-)], C [(C6H5)(C6H5)], D [(C7H6+)(C7H6+)], and E [(C8H72+)(C8H72+)] (Bond length in Å, molecules A, D, E (DA = 90°), molecule B (DA = 30°), molecule C (DA = 50°)
Delocalized Bond Energy (EDBE) is used to describe the total ring bonds energy of the molecule in which all ring bonds are equal and the electrons are delocalized[8]. The ratio of EBLE and EDBE reflects the percentage of the decreased energy.
EDBE is also a difference between two Single Point Energies, which is similar to EBLE, and it could be calculated according to formula (1) with the method of B3LYP/6-311+g (d, p) under Gaussian09.
A detailed splitting method of EDBE calculation could be seen in Fig. 3.
Fig. 3a and Fig. 3b are illustrations showing how to calculate EDBE of [(C3H2+)(C3H2+)] and [(C5H4-)(C5H4-)], respectively. According to the method and above figures, we can calculate the EDBE of [(C6H5)(C6H5)], [(C7H6+)(C7H6+)] and [(C8H72+)(C8H72+)]. And Fig. 3c gives us a more detailed operation to calculate the EDBE of DHRM [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5, 6, 7, 8, m = +1, –1, 0, +1, +2), where 2s+1 is the multi spin of the debris. The multi spin and single point energy of debris C-H will be different because of its electric charge. A higher value of multi spin corresponds to a higher energy of the debris. The detailed value of [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge) will be displayed in the following context.
2.2.3
Calculation of degree of aromaticity (DOA)
Degree of Aromaticity (DOA), a two-dimensional intensive quantity including geometric and energetic factors, as a new measurement of aromaticity has been proposed recently from our group[8, 12]. Only when these three quantities of Ring radius, Bond Length Equalization energy EBLE, and Delocalized Bond Energy EDBE were displayed can we produce the value of DOA.
The proposed formula of DOA is:
$ \mathrm{DOA}=\frac{\mathrm{E}_{B L E}}{\mathrm{E}_{D B E} * \mathrm{r}^2}$
(2)
The value of DOA of [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5, 6, 7, 8, m = +1, –1, 0, +1, +2) will be displayed in Section 3.
3.
RESULTS AND DISCUSSION
3.1
Optimized structure of DHRM
Because each ring of DRHM is planar, there are four quantities to adjust the structure of DHRM: radius of each ring, dihedral angle (DA) of the two ring planes, bond length of the bridge to connect two rings of DHRM, and bond length of G-H, which are all displayed in Table 1.
Table 1
Table 1.
Ring Radius r (Å), Bridge Bond Lengths of the Two Rings BD (Å), Dihedral Angle DA (°), EBLE (kcal/mol) and EDBE (kcal/mol) of DHRM [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2)
Optimized structure is the lowest energy structure of each DHRM.
D2d is the molecular point group of DRHM[(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2) when the dihedral angle of the two rings is 90°.
The following Potential Energy Surface (PES) scan based on optimized structure tells us that the DA of the two rings is important to determine the structure of DHRM. As n = 3, 7 and 8, [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge) fall to its lowest molecular energy when DA = 90° without any imaginary frequency; while n = 5, 6 and DA = 90°, [(GnHn–1m)(GnHn–1m)] is not a stable structure but merely a transition state, and has an imaginary frequency.
PES scan was performed to determine the optimal DA of the two rings with the method of Hartree Fock, and the basis set of 3-21g is precise enough for DA determining and time saving.
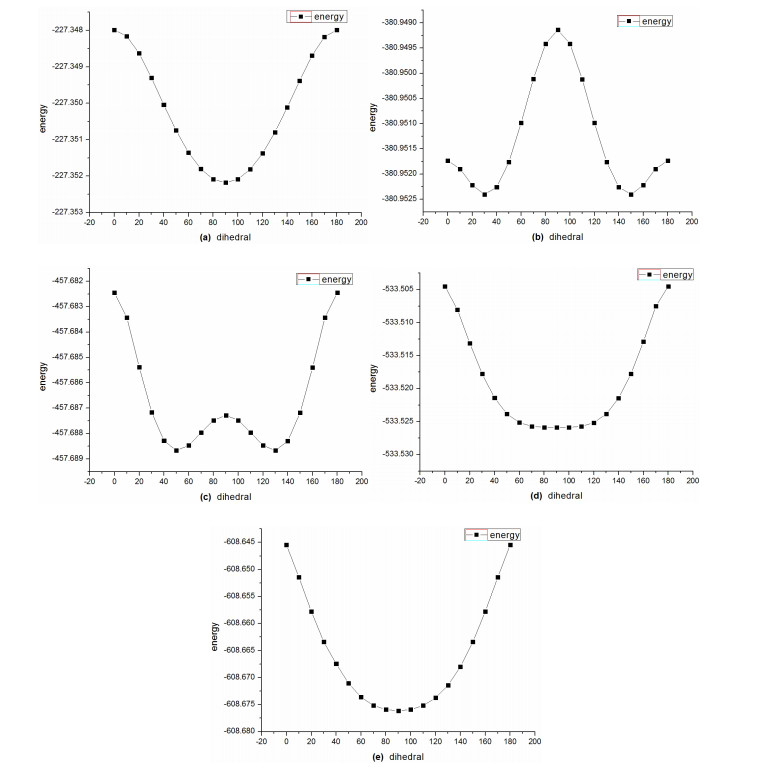
From Fig. 5 we can see DA = 90° is the local minimum of DRHM (n = 3, 7 and 8), the first derivative of the Energy versus DA is equal to 0, and the second one is greater than 0. When n = 5 and 6, DA = 90° is not the local minimum of DRHM, even though it is a maximum ranging from 0° to 180° because the density of its π electron is greater than DRHM (n = 3, 7, 8).
Figure 5
Figure 5.
PES scan of the dihedral angle (DA) of the two ring planes of DRHM (a) [(C3H2+)(C3H2+)], (b) [(C5H4-)(C5H4-)], (c) [(C6H5) (C6H5)], (d) [(C7H6+) (C7H6+)] and (e) [(C8H72+) (C8H72+)] from 0° to 180° at an interval of 10°
DA = 30° is the local minimum of DRHM (n = 5), which was proved again with the method of B3LYP/6-311G+ (d, p) when all other quantities are optimal except the DA. The energy difference between the two conformations, DA = 30° and 90°, is 1.68 kcal/mol, and the structure loses its imaginary frequency from DA = 30° to DA = 90°.
DA = 50° is the local minimum of DRHM (n = 6) which was proved again with the B3LYP/6-311G+ (d, p) method when all other quantities are optimal except the DA. The energy difference between the two conformations DA = 50° and DA = 90° is 1.89 kcal/mol, and the structure loses its imaginary frequency from DA = 50° to DA = 90°.
So the optimal DA are 90° (n = 3, 7, 8), DA = 30° (n = 5) and 50° (n = 6). The molecular symmetry of DRHM (DA = 30° or 50 °) is D2 lower than D2d.
3.2
Electronic structure of DHRM
The lowest energy point is the stable structure of a molecule. For DHRM[(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2), different DAs correspond to different molecular energies, but why molecular energy changes depending on DA, and why DA = 90° is the stable point where n = 3, 7, 8 rather than n = 5 and n = 6. We can get some comprehension from its electronic structure[2].
DA = 0° isn't the lowest energy point of [(CnHn–1m)(CnHn–1m)] (n = 3, 5~8, m = +1, –1, 0, +1, +2), because one ring with the number of π electrons to be 4n+2 is aromatic, while both two rings with the π electrons of 4n are anti-aromatic. And an anti-aromatic structure corresponds to a higher molecular energy.
From Fig. 6, we can see the electronic cloud of [(C6H5)(C6H5)].
Figure 6
Figure 6.π Bonding molecular orbital and π anti-bonding molecular orbital of dihedral angle of [(C6H5)(C6H5)] as 0°, 50° and 90°, respectively
DA = 0°, HOMO-6 is an anti-bonding molecular orbital from the region of global, even though there are two local π bonding orbitals, but its total energy isn't the lowest; HOMO-9 is a bonding orbital, but its π electrons are 12 and it is anti-aromatic, so its energy isn't the lowest as well.
DA = 90°, HOMO-7 and HOMO-8 have only one π bonding orbital respectively. The delocalization of π electrons lowers the molecular energy, but the other ring is anti-bonding and increases the energy, so its energy isn't the lowest as well.
DA = 50° is a transitional structure between DA = 0° and DA = 90°, so it takes both advantages of them and becomes the stable point of [(C6H5)(C6H5)].
3.3
EBLE and EDBE of DHRM
Based on the stable structure of quantities in Table 1 and the method mentioned above, we can calculate the EBLE, EDBE and DOA of DHRM[(GnHn–1m)(GnHn–1m)], and then further study the aromaticity of DHRM which is performed in one ring of DHRM because the rings are symmetric with each other. The methods to calculate EBLE and EDBE have been given previously, with the detailed data listed in Table 1.
From Table 1, we can see the value of EBLE increases as the number of ring atoms increases from 3 to 8. As the ring atoms increase, the ring radius is increased gradually. EBLE increases with n increasing from 3 to 8 of [(GnHn–1m)(GnHn–1m)] as long as the element of G doesn't change. The value of EBLE ranges from 10 to 40 kcal/mol which is a same magnitude with the aromatic stabilization energy measured in the experiments[9].
The values of EDBE are given in Table 1 with the magnitude being 102 kcal/mol which reflects the amounts of bond energy of the ring. EDBE increases as the number of ring atoms increases with an interval of 200~300 kcal/mol.
Different from the energy interval where n = 3, 5 and 6, EDBE (n = 7) approximates to EDBE (n = 8), because debris energy of CH+ is higher than the energy of CH by about 250 kcal/mol. Moreover, DHRM (n = 8) has two CH+ debris while DHRM (n = 7) has only one. Another point is that the decreased energy in the process of bond length equalization only takes a small occupation of ring bond energy when comparing the values of EBLE and EDBE of a same molecule.
3.4
Aromaticity of DHRM
DOA and NICS will be the major reference to study the aromaticity of DHRMs. Similarities and differences between DHRM and its corresponding single ring molecules of DOA and NICS will also be considered.
3.4.1
DOA of DHRM
For a series of DHRMs of one element, the values of DOA are decreasing with increasing the number of cyclization atoms, namely the aromaticity is decreased gradually. The interval of DOA among DHRMs (n = 5, 6, 7 and 8) is smaller than the interval between n = 3 and 5; because the number of π electrons is all six (n = 5, 6, 7, 8), and that of π electrons is all two (n = 3), it is noteworthy that the radius of DHRM (n = 3) is very shorter than DHRM (n = 5, 6, 7, 8).
The difference of DOAs between DHRM and its corresponding single ring molecule is small, and they located on the same magnitude order. The property of DOA keeps its continuity from single ring molecule to its corresponding DHRM.
From Table 2 we can see that the ring radius of DHRM is greater than that of its corresponding single ring molecule in general, and because the former has more atoms than the latter, its greater repulsion of more atoms increases its ring radius to keep the mechanical equilibrium. Increasing the ring radius leads to a smaller DOA according to the formula of DOA.
Table 2
Table 2.
DOA (Å–2) of DHRM [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2) and Its Corresponding Single Ring Molecules and Their r (Å)
Schleyer and co-workers proposed the use of absolute magnetic shieldings, calculated at ring centers with available quantum packages, called Nucleus-Independent Chemical Shift (NICS), as a new aromaticity criterion[19]. NICS became popular because it can be easily calculated to characterize the aromatic degree of organic molecules[2], the inorganic aromatic molecules[2], all metal series[14] and sandwich aromatic matching molecules[2~4]. Significantly negative NICS values denote its diatropic ring currents and aromaticity, whereas positive values show its paratropic ring currents and antiaromaticity. The zero NICS value means no-aromaticity[2].
Table 3 shows that for a series of DHRMs of one element, the values of NICS decrease with increasing the number of cyclization atoms, namely the aromaticity is increased gradually because a more negative NICS corresponds to a bigger aromatic degree.
Table 3
Table 3.
NICS(-ppm) of [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2) and Its Corresponding Single Ring Molecules
Owing to the little change of its chemical and magnetic environment, only one of the hydrogen atoms was replaced by another ring, so the difference of NICS between DHRM and its corresponding single ring molecule is small.
3.4.3
Correlation between DOA and NICS of DHRMs
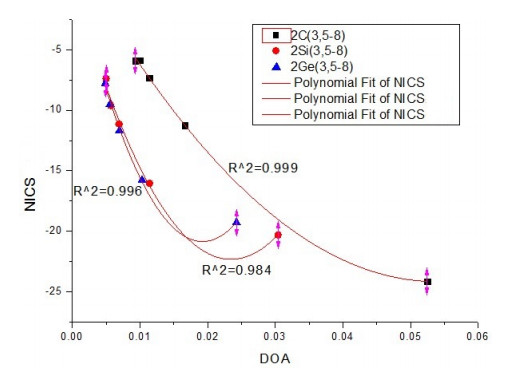
We plot to fit the correlation between DOA and NICS of DHRMs, where DOA is the horizontal axis and NICS is the vertical axis, to analyze the aromaticity of DHRM further.
From Fig. 7 we can see, for a series of DHRM of one element (C or Si or Ge), a more negative NICS corresponds to a bigger DOA, namely a bigger aromaticity. Fitted curve shows the quadratic correlation between DOA and NICS which is similar to the correlation of its corresponding single ring molecules. The correlation with NICS reflects the validity of DOA.
Figure 7
Figure 7.
Correlation between DOA and NICS of DHRM, R^2 (correlation coefficient)
From the above correlation equations we can see that the constant coefficient, linear coefficients and quadratic coefficients of these three equations were located on the same number order respectively. The linear and quadratic coefficients of the three functions increased gradually with the order of C, Si and Ge, which was a same order of the increasing atomic mass.
According to the data of Tables 3 and 2, for too many DHRMs and its corresponding single ring molecule, a more negative NICS corresponds to a bigger DOA, namely a bigger aromaticity, such as (C5H5)-, its (DOA, NICS) = (0.01752, –12.6), and its corresponding DHRM [(C5H4)-(C5H4)-]2-, and its (DOA, NICS) = (0.01662, –11.3). Although there was no accurate relationship of size between DOA and NICS, DOA could be an indication of aromaticity just as NICS does because they have a rough relationship of size.
3.5
RSVRSF of DHRM
Ring Stretching Vibration Raman Spectroscopy Frequency (RSVRSF) of single ring molecules is an effective aromatic probe[5]. A bigger RSVRSF corresponds to a bigger aromaticity. What is the relation between RSVRSF and aromaticity among DHRMs? RSVRSF and its intensity of DHRMs [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2) were performed via Gaussian09 with the method of B3LYP/6-311+g (d, p) based on its optimized structure.
From Table 4 we can see, for a series of DHRMs of one element (C or Si or Ge), the value of RSVRSF decreases gradually as the number of cyclization atoms increases, which shows the same tendency with its corresponding single ring molecules. The value of RSVRSF of DHRM is very close to that of its corresponding single ring molecules, which could act as the characteristic frequency of ring molecules to identify its aromaticity and vibration mode.
Table 4
Table 4.
RSVRSF (s–1) and Its Intensity of DHRM and Its Corresponding Single Ring Molecules
3.6
Correlation of RSVRSF vs. DOA and RSVRSF vs. NICS of DHRMs
3.6.1
Correlation between RSVRSF and DOA of DHRMs
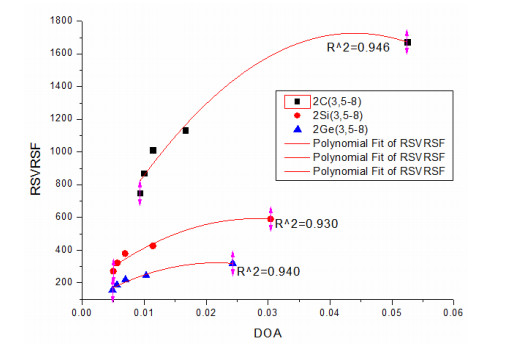
In order to study the correlation between RSVRSF and DOA, we plot the fitting curve where DOA is the horizontal axis and RSVRSF is the vertical axis, as shown in Fig. 8.
Figure 8
Figure 8.
Correlation between DOA and RSVRSF of DHRMs
Fig. 8 shows that for a series of DHRM of one element (C or Si or Ge), a bigger RSVRSF corresponds to a bigger DOA, namely a higher aromaticity. Fitted curve shows a quadratic correlation between DOA and RSVRSF which was similar to the correlation of its corresponding single ring molecules.
Taking DOA as x-coordinate and RSVRSF as y-coordinate, the respective correlation equations of DHRM are as below:
From the above correlation equations we can see, the constant coefficient, linear coefficients and quadratic coefficients of these three equations were located on the same number order respectively. All of the three coefficients of these equations decreased gradually with the order of C, Si and Ge, which was an opposite order of increasing the atomic mass.
According to the data of Tables 2 and 4, for most ring molecules of DHRMs and its corresponding single ring molecules, a bigger RSVRSF corresponds to a bigger DOA, namely a larger aromaticity, such as (C5H5)-, its (DOA, RSVRSF) = (0.01752, 1143.5), and its corresponding DHRM [(C5H4)-(C5H4)-], and its (DOA, RSVRSF) = (0.01662, 1134.0). They have a rough relationship of size between RSVRSF and DOA, which proves again that RSVRSF is also an aromatic probe among DHRMs.
3.6.2
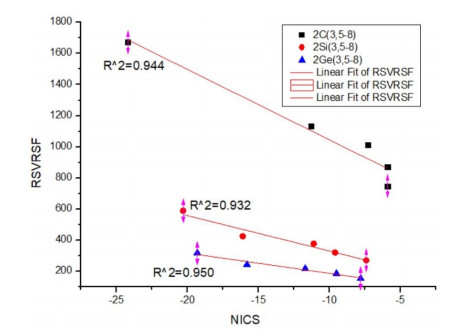
Correlation between RSVRSF and NICS of DHRM
In order to study the correlation between RSVRSF and NICS, we plot the fitting curve taking NICS as the horizontal axis and RSVRSF as the vertical axis (Fig. 9).
Figure 9
Figure 9.
Correlation between RSVRSF and NICS of DHRMs
From the above correlation equations we can see that both of the constant and linear coefficients of these three equations were located on the same number order respectively. Both of the two coefficients of the three equations decreased gradually with the order of C, Si and Ge, which was an opposite order of increasing the atomic mass. The value of RSVRSF decreases as the order of C > Si > Ge, if the number of ring atoms of the two DHRMs is equal; and the value of absolute value of NICS (0, 0) keeps the same order (C > Si > Ge) with RSVRSF.
4.
CONCLUSION
Dihedral Angle (DA) is an important factor for the search of optimal structure of DHRM [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2); DA = 90° is the stable structure when n = 3, 7 and 8; while n = 5 corresponds to DA = 30°, and n = 6 to DA = 50°.
For a series of DHRMs of one element (C or Si or Ge), the value of DOA decreases with the increasing number of cyclization atoms, and the aromaticity is decreased gradually. The property of DOA keeps its continuity from single ring molecule to its corresponding DHRM. Absolute value of NICS decreases with increasing the number of cyclization atoms, and the aromaticity drops gradually. The value of RSVRSF decreases gradually with the increasing number of cyclization atoms, which shows a same tendency with its corresponding single ring molecules. The value of RSVRSF of DHRM is very close to that of its corresponding single ring molecules, which could act as characteristic frequency of ring molecules to identify its aromaticity and vibration mode.
The correlation between DOA and NICS of DHRMs is quadratic, which reflects the validity of DOA. Correlation between RSVRSF and DOA of DHRMs is quadratic; correlation between its RSVRSF and NICS is linear, which shows a same tendency with its corresponding single ring molecules.
[1]
Kruszewski, J.; Krygowski, T. M. Definition of aromaticity basing on the harmonic oscillator model. Tetra. Lett.1972, 13, 3839−3842. doi: 10.1016/S0040-4039(01)94175-9
[2]
Mandal, D.; Maity, R.; Beg, H.; Salgado-M, G.; Misra, A. Computation of global reactivity descriptors and first hyper polarisability as a function of torsional angle of donor-acceptor substituted biphenyl ring system. Mol. Phys. 2017, 116, 1−11.
[3]
Wei, X. Y.; Zhao, B. X.; Shang, Y. Z.; Cheng, Y. Z. Rigid biphenyl-contained epoxy resins with improved thermal resistant properties. Chin. J. Poly. Sci.2017, 35, 1428−1435. doi: 10.1007/s10118-017-1975-9
[4]
Boffetta, P.; Catalani, S.; Tomasi, C.; Pira, E.; Apostoli, P. Occupational exposure to polychlorinated biphenyls and risk of cutaneous melanoma. Eur. J. Cancer Prev.2018, 27, 62−69. doi: 10.1097/CEJ.0000000000000316
[5]
Tanabe, Y.; Mizuhata, Y.; Tokitoh, N. Synthesis and structure of a heavier congener of biphenyl, 1, 1΄-disila-4, 4΄-biphenyl. Organometallics2010, 29, 721−723. doi: 10.1021/om901095w
[6]
Guo, Y. B.; Liu, Z. Z.; Liu, H. X.; Zhang, F. Y.; Yin, J. Q. A new aromatic probe — the ring stretching vibration Raman spectroscopy frequency. Spectrochim. Acta A2016, 164, 84−88. doi: 10.1016/j.saa.2016.03.005
[7]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Wong, M. W.; Foresman, J. B.; Robb, M. A.; Head-Gordon, M.; Replogle, E. S.; Gomperts, R.; Andres, J. L.; Raghavachari, K.; Binkley, J. S.; Gonzalez, C.; Martin, R. L.; Fox, D. J.; Defrees, D. J.; Baker, J.; Stewart, J. J. P.; Pople, J. A. Gaussian 92/DFT
[8]
Shen, C. F.; Liu, Z. Z.; Liu, H. X.; Zhang, H. Q. Bond length equalization with molecular aromaticity-a new measurement of aromaticity. Spectrochim. Acta A2018, 201.
[9]
Cyranski, M. K. Energetic aspects of cyclic pi-electron delocalization, evaluation of the methods of estimating aromatic stabilization energies. Chem. Rev.2005, 5, 3773−3811.
[10]
Aihara, J. Effect of bond-length alternation on the aromaticity of benzene. Bull. Chem. Soc. Jpn.1990, 63, 1956−1960. doi: 10.1246/bcsj.63.1956
[11]
Aihara, J. Bond-length equalization and aromaticity in charged π-systems. Bull. Chem. Soc. Jpn.2004, 77, 2179−2183. doi: 10.1246/bcsj.77.2179
[12]
Solomon, P. M.; Norton, D. L. The biphenyl molecule as a model transistor. ACS Nano. 2008, 2, 435−440. doi: 10.1021/nn700253p
[13]
Jiao, H.; Schleyer, P. V. R.; Mo, Y.; McAllister, M. A.; Tidwell, T. T. Magnetic evidence for the aromaticity and antiaromaticity of charged fluorenyl, indenyl, and cyclopentadienyl systems. J. Am. Chem. Soc.1997, 119, 7075−7083. doi: 10.1021/ja970380x
[14]
Mercero, J. M.; Ugalde, J. M. Sandwich-like complexes based on "all-metal" (Al 4 2-) aromatic compounds. J. Am. Chem. Soc.2004, 126, 3380−3381. doi: 10.1021/ja039074b
[15]
Liu, Z. Z.; Tian, W. Q.; Feng, J. K.; Zhang, G.; Li, W. Q. Theoretical study on structures and aromaticities of P5-anion, [Ti(η5-P5)]- and sandwich complex [Ti(η5-P5)2]2. J. Phys. Chem. A2005, 109, 5645−5655. doi: 10.1021/jp044395a
[16]
Liu, Z. Z.; Tian, W. Q.; Feng, J. K.; Zhang, G.; Li, W. Q. The electronic structures and aromaticities for zinc sandwich, half-sandwich and zinc-zinc (Zn22+) sandwich complexes within density functional theory. J. Mol. Struc. Theochem. 2006, 758, 127−138. doi: 10.1016/j.theochem.2005.10.025
[17]
Liu, Z. Z.; Tian, W. Q.; Feng, J. K.; Zhang, G.; Li, W. Q. A theoretical study on structures, bonding energies and aromaticity of two new series of dinuclear phosphametallocenes, (η5-P5) MM΄(η5-P5) and (η5-C5H5) MM΄(η5-P5) (M, M΄ = Zn, Cd). Eur. J. Inorg. Chem. 2006, 14, 2808−2818.
[18]
Liu, Z. Z.; Tian, W. Q.; Feng, J. K.; Zhang, G.; Li, W. Q. Theoretical study on structures and aromaticities of a new series of sandwich complexes, [M2(η5P5)2] and [M(η5P5)2] (M = Be, Mg, and Ca). J. Mol. Struct. Theochem. 2007, 809, 171−179. doi: 10.1016/j.theochem.2007.01.020
[19]
Schleyer, P. V. R.; Maerker, C.; Dransfeld, A.; Jiao, H. J.; Hommes, N. J. R. V. E. Nucleus-independent chemical shifts, a simple and efficient aromaticity probe. J. Am. Chem. Soc.1996, 118, 6317−6318. doi: 10.1021/ja960582d
Figure 1
An illustration of C6H6 in the process of BLE with its structure from left to right. Ring radius r, single bond radius angle $ {\mathit{\theta }}_{1} $, double bond radius angle $ {\mathit{\theta }}_{2} $, single bond length $ {\mathit{B}\mathit{L}}_{1} $, double bond length $ {\mathit{B}\mathit{L}}_{2} $
Figure 2
An illustration of EBLE of DHRM [(CnHn-1m)(CnHn-1m)] (Right rings are single and double ring bonds alternatively, and left rings are all equal ring bonds. It should be compared with Fig. 4)
Figure 3a
Splitting structure illustration of EDBE of [(C3H2+)(C3H2+)] (Violet atoms are the dummy atoms that are only helpful for comprehending, but make no difference for the results)
Figure 4
Optimized structures of DHRM, A [(C3H2+)(C3H2+)], B [(C5H4-)(C5H4-)], C [(C6H5)(C6H5)], D [(C7H6+)(C7H6+)], and E [(C8H72+)(C8H72+)] (Bond length in Å, molecules A, D, E (DA = 90°), molecule B (DA = 30°), molecule C (DA = 50°)
Figure 5
PES scan of the dihedral angle (DA) of the two ring planes of DRHM (a) [(C3H2+)(C3H2+)], (b) [(C5H4-)(C5H4-)], (c) [(C6H5) (C6H5)], (d) [(C7H6+) (C7H6+)] and (e) [(C8H72+) (C8H72+)] from 0° to 180° at an interval of 10°
Table 1.
Ring Radius r (Å), Bridge Bond Lengths of the Two Rings BD (Å), Dihedral Angle DA (°), EBLE (kcal/mol) and EDBE (kcal/mol) of DHRM [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2)
Table 2.
DOA (Å–2) of DHRM [(GnHn–1m)(GnHn–1m)] (G = C, Si, Ge, n = 3, 5~8, m = +1, –1, 0, +1, +2) and Its Corresponding Single Ring Molecules and Their r (Å)

 DownLoad:
DownLoad:





 下载:
下载:












