引用本文:
陈祖华, 方圆, 吴波, 吴育锋, 范志宇, 萨百晟, 郑福南, 黄锦涛. AxMn1-xFe2O4铁氧体 (A=Zn,Ni) 中阳离子的占位有序化行为研究[J]. 无机化学学报,
2016, 32(3): 377-385.
doi:
10.11862/CJIC.2016.026

Citation: CHEN Zu-Hua, FANG Yuan, WU Bo, WU Yu-Feng, FAN Zhi-Yu, SA Bai-Sheng, ZHENG Fu-Nan, HUANG Jin-Tao. Study of the Site Occupancy Ordering Behaviors of Cations in the AxMn1-xFe2O4 Ferrites (A=Zn, Ni)[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(3): 377-385. doi: 10.11862/CJIC.2016.026
Citation: CHEN Zu-Hua, FANG Yuan, WU Bo, WU Yu-Feng, FAN Zhi-Yu, SA Bai-Sheng, ZHENG Fu-Nan, HUANG Jin-Tao. Study of the Site Occupancy Ordering Behaviors of Cations in the AxMn1-xFe2O4 Ferrites (A=Zn, Ni)[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(3): 377-385. doi: 10.11862/CJIC.2016.026
AxMn1-xFe2O4铁氧体 (A=Zn,Ni) 中阳离子的占位有序化行为研究
摘要:
基于尖晶石晶体结构信息,本文采用热力学三亚晶格模型,将材料热力学计算和第一性原理计算相结合,研究了ZnxMn1-xFe2O4和NixMn1-xFe2O4立方相中的Zn2+、Ni2+、Mn2+以及Fe3+在8a和16d亚晶格上的占位有序化行为。结果表明:在锰铁氧体中,室温下Mn2+完全占据在8a亚晶格上,Fe3+完全占据在16d亚晶格上,属于正尖晶石结构;随着热处理温度升高,在1 273 K达到热处理平衡时的占位构型为 (Fe0.093+Mn0.912+)[Fe1.913+Mn0.092+]O4,在热处理温度升至1 473 K时,达到热处理平衡时的占位构型为 (Fe0.113+ Mn0.892+)[Fe1.893+Mn0.112+]O4,均与实验结果符合较好。在锌铁氧体中,室温下Zn2+完全占据在8a亚晶格上,Fe3+完全占据在16d亚晶格上,属于正尖晶石结构;在热处理温度较高时,Zn2+和Fe3+发生部分置换,符合实验结果。在镍铁氧体中,半数的Fe3+在室温下占据在8a亚晶格上,Ni2+与剩下另一半的Fe3+共同占据在16d亚晶格上,仅在热处理温度较高的时候发生微弱变化,亦与已有的实验结果吻合。在此基础上,本文进一步通过热力学模型研究了立方相尖晶石结构的ZnxMn1-xFe2O4、NixMn1-xFe2O4复合体系中阳离子占位行为与热处理温度对占位的影响规律。
English
Study of the Site Occupancy Ordering Behaviors of Cations in the AxMn1-xFe2O4 Ferrites (A=Zn, Ni)
Abstract:
Based on the crystal structure of spinel, we applied the thermodynamic ternary sublattices model to investigate the site occupancy ordering behaviors of Zn2+, Ni2+, Mn2+ and Fe3+ on the 8a and 16d sublattices in the ZnxMn1-xFe2O4 and NixMn1-xFe2O4 by combining thermodynamic calculations with first-principles calculations. Our results suggest that in the manganese ferrites, Mn2+ occupies the 8a sublattice completely, and Fe3+ occupies the 16d sublattice at room temperature, which belongs to a normal spinel. The site configuration is (Fe0.093+Mn0.912+) [Fe1.913+Mn0.092+]O4, when the ferrite is subjected to the heat treatment of 1 273 K. When the temperature increases to 1 473 K, the site configuration is (Fe0.113+Mn0.892+)[Fe1.893+Mn0.112+]O4, which agrees well with the available reliable experimental results. In the zinc ferrites, Zn2+ occupies the 8a sublattice completely, and Fe3+ completely occupies the 16d sublattice at room temperature. It also belongs to normal spinel, and the cations trend to exchange the site occupancy each other at higher temperature. In the Ni ferrites, all Ni2+ occupies the 16d sublattice, and the Fe3+ cations occupy both the 8a and the 16d sublattices, which agrees with the experimental results. It belongs to an inverse spinel. Then, the relationship between the occupancy behaviors and the composition, as well as the heat treatment temperature in the ZnxMn1-xFe2O4 and NixMn1-xFe2O4 were established based on theoretically predictions.
-
Key words:
- spinel
- / Mn-Zn ferrites
- / ordering behavior
- / thermodynamic model
- / first-principles calculations
-
0 引言
软磁铁氧体是软磁材料的典型代表[1],由于其具有高磁导率、高饱和磁感强度、低矫顽力和低损耗等优点,因此,被广泛用于各种电感元件如滤波器磁芯、变压器磁芯、天线磁芯、磁带录音和录像磁头以及多路通讯等的记忆磁头磁芯等,在电子工业中的应用具有悠久历史,并且日益广泛[2-3]。随着电子信息技术的飞速发展,电子设备趋向集成智能化、小型轻量化发展,对高性能软磁铁氧体材料的要求也越来越高。因此,从理论上弄清软磁铁氧体的微结构和性能[4-6],有助于加速高性能低成本软磁铁氧体的研究开发。
众所周知,离子或原子在亚晶格上的占位行为对材料的性能具有重要的影响。由于制备样品的粒径尺寸效应和热处理工艺的影响[7],尖晶石结构的铁氧体化合物中阳离子的分布具有多样性。因此,对尖晶石结构的铁氧体化合物中阳离子在亚晶格上的占位有序化行为的研究结果也具有一定的差异性[8-9]。在理论研究方面,Seko等采用团簇扩张法结合第一性原理计算的方法,研究了六种不同成分的尖晶石氧化物[10],其研究结果表明MgGa2O4和MgIn2O4是四方相的反式尖晶石结构,而MgAl2O4,ZnAl2O4,ZnGa2O4和ZnIn2O4是立方相的正尖晶石结构。Wei等用反置能量 (Inversion Energy) 来判断某一体系的尖晶石结构化合物是属于正尖晶石结构还是反尖晶石结构[11],并且基于SQS (Special Quasira-ndom Structure) 近似方法,具体研究了AⅡB2ⅢO4和AⅣB2ⅡO4系列尖晶石结构中的阳离子分布,其中研究的元素包括第Ⅱ主族中的Mg、Zn和Cd元素,第Ⅲ主族中的Al、Ga、和In元素, 以及第Ⅳ主族中的Si、Ge和Sn元素。Wei等采用XRD分析方法研究了在1 523 K下制备的NixMn1-xFe2O4铁氧体中阳离子的占位分布情况,并采用热力学计算的方法,分析了1 523 K下该体系铁氧体中阳离子的占位有序化行为,其与实验结果吻合得较好[12]。但是,其热力学推导过程在建模时需要基于反应平衡常数等一些实验参数,并且,它假设Ni在8a亚晶格上的占位分数为0,这些都是依赖于实验与经验。
本文基于立方相尖晶石的晶体结构信息,通过建立热力学亚晶格模型,辅助以第一性原理计算,来定量化研究AxMn1-xFe2O4铁氧体A=Zn,Ni) 中阳离子在亚晶格上的占位有序化行为随成分和温度的变化规律,以期为AxMn1-xFe2O4铁氧体 (A=Zn,Ni) 软磁材料的配方设计和性能调控提供理论指导。
1 计算模型和计算方法
1.1 计算模型
本文研究立方相尖晶石结构的铁氧体中阳离子在亚晶格上的占位有序化行为,其晶体结构信息如表 1所示,其单胞结构如图 1所示。该结构为立方结构,其中氧离子构成面心立方密堆垛结构,共有64个四面体间隙和32个八面体间隙。关于尖晶石结构中,离子的等效Wyckoff位置和内坐标参数的选取和转换方法,Sickafus等进行了详细的介绍[13],根据阴离子坐标u的对称性,当u的对称性分别为-43m和-3m时,分别对应于两种亚晶格模型,即8a、16d、32e和8b、16c、32e。在完全有序的锰铁氧体尖晶石结构中,本研究将Mn2+填充在其1/8的四面体间隙中,用Wyckoff晶位8a表示;Fe3+填充在1/2的八面体间隙中,用Wyckoff晶位16d表示;而氧离子Wyckoff晶位用32e表示。基于上述3套Wyckoff亚晶格位置,即8a、16d、32e,本文建立出立方相尖晶石结构铁氧体的亚晶格模型。
 表 1
立方相尖晶石结构铁氧体的晶体学结构信息
Table 1.
Spinel (MgAl2O4-H11) Structure
表 1
立方相尖晶石结构铁氧体的晶体学结构信息
Table 1.
Spinel (MgAl2O4-H11) Structure
Prototype Pearson's symbol Space Group Sublattice Atomic Coordinate MgAl2O4 cF56 Fd3m 227 Multiplicity Wyckoff notation Point symmetry x y z 8 a -43m 0 0 0 l6 d -3m 0.625 0.625 0.625 32 e 3m 0.375 0.375 0.375 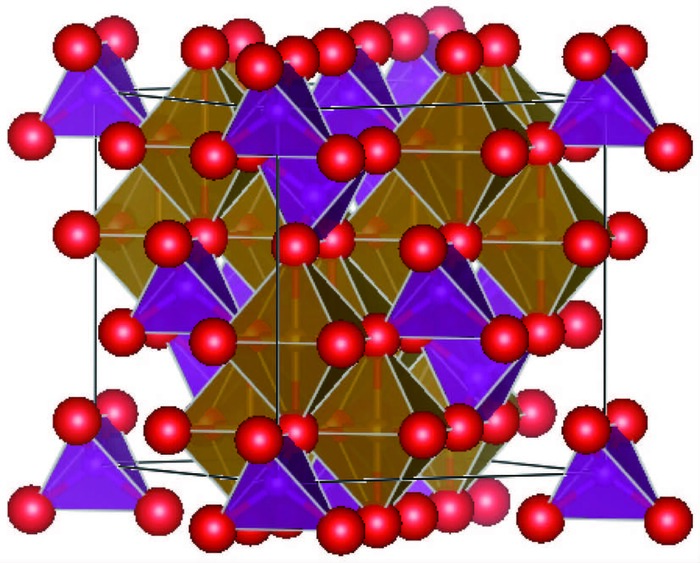 图 1
MnFe2O4的晶体结构
Figure 1.
Crystal structure of MnFe2O4
图 1
MnFe2O4的晶体结构
Figure 1.
Crystal structure of MnFe2O4
本文基于晶体学结构信息,采用亚晶格模型研究铁氧体中阳离子在亚晶格上的占位有序化行为。亚晶格模型,也称为化合物能量模型 (Compound Energy Formalism)[14],可以用于预测化合物中各组元在各个亚晶格中的占位分数,进而研究元素的占位有序化行为。亚晶格模型可以用表达式 (1) 表示:
其中,
$y_{{{E}_{i}}}^{{{W}_{i}}}$ 表示合金元素Ei在亚晶格Wi(以Wyckoff位置表示) 中的占位分数;${{f}_{{{W}_{i}}}}$ 表示亚晶格Wi的等效位置个数在所有点阵中所占的比例。该模型不受组元数、亚晶格个数的限制,能够对任意复杂体系的晶体构型和热力学进行有效的描述。对于立方相尖晶石结构铁氧体,W1=8a、W2=16d和W3=32e。根据文献报道,氧元素完全占据在32e亚晶格上,因此本文将氧离子固定在32e亚晶格上,以锰锌铁氧体为例,金属阳离子的占位模型如表达式 (2) 所示:
根据热力学条件,由室温下稳定结构的纯组元形成化合物的Gibbs自由能如公式 (3) 所示:
其中,ΔH为生成焓,ΔS为形成熵,T为温度。由于直接求解ΔG是困难的,本文根据热力学状态函数的固有性质,即热力学量的大小只与始末态有关,而与中间过程无关的这一特性,设计一条等效路径来间接计算出化合物的Gibbs自由能。根据热力学亚晶格模型,定义一系列端基化合物[14],即某一种点阵上只被一种元素占据,根据排列,m种阳离子在n种亚晶格上的占据,其排列端基数为mn。由于氧离子的占位是确定的,仅需考虑体系中阳离子在8a和16d两种亚晶格上的占位行为。因此,对于锰锌铁氧体,m=3,n=2。借助于端基化合物这一中间过程,将元素形成化合物的过程,分解为由元素先形成端基化合物,然后再由端基化合物组装形成复杂化合物这样一个等效的替换途径来间接计算出化合物的Gibbs生成自由能,计算转换途径如图 2所示。
 图 2
热力学函数计算路径转换示意图 (其中反应路径③ 可以转换为由路径① 和路径② 联合来实现,即③=①+②)
Figure 2.
Alternative computing path of thermodynamic function denoting ③=①+②
图 2
热力学函数计算路径转换示意图 (其中反应路径③ 可以转换为由路径① 和路径② 联合来实现,即③=①+②)
Figure 2.
Alternative computing path of thermodynamic function denoting ③=①+②
因此,ΔH的表达如公式 (4) 所示:
其中,ΔH(k:l:m)为室温稳定结构的纯组元生成端基化合物的生成焓,ΔEH为由室温稳定结构的纯组元生成锰锌铁氧体的过剩焓,由于过剩焓的值很小,因此本文不予考虑[15]。
Jain等指出,温度和压力对固相材料的热容影响非常微弱[16],而且Lany等计算出来的ΔH298 K和ΔH0 K差值一般小于0.03 eV·atom-1[17], 因此本文只考虑0 K下的生成焓,并由公式 (5) 计算得到,公式 (5) 为:
其中,ΔH(k:l:m),Etot (k),Etot (l)和Etot (m)分别为端基化合物和相应纯组元的总能,其单位为eV·atom-1。
同理,忽略由室温稳定结构的纯组元生成锰锌铁氧体的过剩熵,将化合物的生成熵近似简化为体系的混合熵[18-19],即如公式 (6) 所示:
体系的混合熵如公式 (7) 所示:
其中,R为波尔兹曼常数,R=8.6194×10-5 eV·K-1·atom-1。将公式 (4) 和 (7) 代入公式 (3),求得体系的Gibbs自由能,其中包含了占位分数与温度两种变量,如公式 (8) 所示:
另一方面,根据质量守恒定律和归一化约束条件,可以得到公式 (9)~(16):
上述各式中,xEi(Ei=Mn,Zn,Fe,O) 表示组元在锰锌铁氧体组成中的物质的量分数。
有序-无序转变达到热力学平衡时,Gibbs自由能最小化,因此根据极值条件获得偏微分方程组,如公式 (17) 所示:
通过第一性原理总能计算 (详见1.2),建立出端基化合物的生成焓热力学数据库后,对于任一给定成分和相平衡温度条件的铁氧体化合物,求解由公式 (9)~(17) 所组成的方程组,即可得到阳离子在各个亚晶格上的占位分数,如公式 (18) 所示:
1.2 计算方法
本文采用基于密度泛函理论的第一性原理计算方法,选用VASP (Vienna Ab-initio Simulation Package)[20]来计算5个纯组元 (Mn、Fe、Ni、Zn、O) 和相应的16个端基化合物的总能,进而获得由纯组元形成各个端基化合物的生成焓。对于每一种端基化合物,均考虑铁磁态,亚铁磁以及反铁磁态这3种状态,并选取总能最低时的状态作为端基化合物的稳定态。交换关联势采用广义梯度近似 (Generalized Gradient Approximation,GGA)[21]的Perdew-Burke-Ernzerh (PBE) 形式来描述,即赝势库文件取PAW-PBE模式。进行总能计算时,对端基化合物,K空间的撒点采用6×6×6 Monkhorst-Pack模式,对室温稳定态的纯组元Mn_BCC的K空间的撒点采用5×5×5 Monkhorst-Pack模式,Fe_BCC、Ni_FCC和Zn_HCP的K空间的撒点均采用12×12×12 Monkhorst-Pack模式,而O2采用4×4×4的Gamma模式,截断能均设置为520 eV。对原子位置、晶胞体积进行弛豫,保持形状仍为立方尖晶石结构。同时,开启自旋极化,初始磁矩依据阳离子的孤对电子数进行设定。计算结果保证总能关于截断动能和K点产生模式的收敛精度达到1 meV·atom-1。
事实上,建立出端基化合物的生成焓热力学数据库后,直接对由公式 (9)~(17) 构成的偏微分方程组进行手工求解难度较大。但是,随着计算材料学的快速发展, 已有专业的热力学计算软件[22], 如Thermo-Calc和Pandat,能够较为方便地求解这类热力学偏微分方程组,并直接给出本文所关注的占位分数。因此,本文在建立好格式化的端基化合物生成焓热力学数据库后,使用Thermo-Calc软件包来计算求解占位分数。
2 结果与讨论
2.1 ZnxMn1-xFe2O4系中的阳离子分布随温度变化的规律
ZnxMn1-xFe2O4体系中阳离子在8a和16d亚晶格上的占位分数随温度的变化如图 3所示,表格2为本文计算值与现有文献值的对照表。
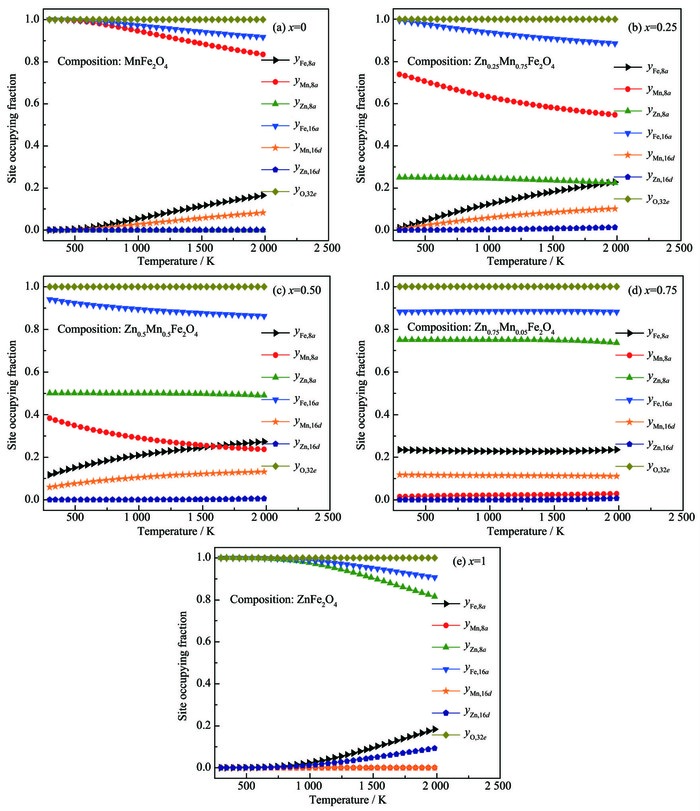 图 3
ZnxMn1-xFe2O4体系立方尖晶石中阳离子的占位分数随热处理温度和成分的变化趋势图
Figure 3.
Cation occupying fraction dependence on heat treatment temperature and composition in cubic spinel ZnxMn1-xFe2O4
图 3
ZnxMn1-xFe2O4体系立方尖晶石中阳离子的占位分数随热处理温度和成分的变化趋势图
Figure 3.
Cation occupying fraction dependence on heat treatment temperature and composition in cubic spinel ZnxMn1-xFe2O4
从图 3可知,当x=0.00时,在室温下Mn2+完全占据在8a亚晶格上,Fe3+完全占据在16d亚晶格上,锰铁氧体的阳离子分布表达式为 (Mn2+)[Fe23+]O4,属于正尖晶石结构,这与Szotek等通过第一性原理的研究,得到锰铁氧体在正尖晶石结构下能量最低,即8a亚晶格完全被Mn元素占据时最稳定[28]的报道相符。另一方面,高温下的占位构型如表 2所示,亦与已有实验结果相符合。当x=0.25时,随着温度的升高,Mn2+逐渐占据在16d亚晶格上,相应地,Fe3+逐渐占据在8a亚晶格上,而Zn2+表现出强烈地占据在8a亚晶格上,但尚未获得可靠实验结果与之对比。本文预测出的Zn2+比Mn2+更加强烈地占据在8a亚晶格上,与Abbas等的研究结果[25]相符合。同时,Henderson等提出,对于大多数的尖晶石结构铁氧体,阳离子在8a和16d亚晶格上不会表现出完全占据在其中一种亚晶格上, 而是表示为 (Mx2+M1-x3+)[M1-x2+M1+x3+]O4[29],这与本文对Zn0.25Mn0.75Fe2O4的阳离子占位分布结果相符合。因此,本文预测结果可以为Zn0.25Mn0.75Fe2O4的占位构型提供理论指导。当x=0.5时,室温下Mn2+与Fe3+即表现了部分反置的占位结果。随着温度的升高,占位有序化程度逐渐减弱,而Zn2+的占位倾向对温度的变化不敏感。当x=0.75时,阳离子的占位分布对温度并不敏感,绝大多数的Mn2+占据在16d亚晶格上,Fe3+迁移到8a亚晶格上,所有的Zn2+依然保持完全占据在8a亚晶格上。从表 2中可以看出,Zn2+和Mn2+与现有文献值差别较大,而Fatemi等指出,Fe3+与Mn2+的离子分布很容易受到第三组元金属阳离子浓度的影响[30],在第三组元金属阳离子浓度较高时,其会发生反置占据。同时,Lee等指出Mn在低浓度的时候倾向于占据在16d亚晶格上,而在浓度较高时倾向于占据在8a亚晶格上[31]。还有实验也表明,Mn2+在低浓度的时候倾向于占据在八面体间隙上,而在高浓度的时候倾向于占据在四面体间隙上[32]。上述文献的占位倾向性研究结果均与本文预测的Mn2+的占位倾向相符合。当x=1时,体系为ZnFe2O4,室温下Zn2+全部占据在8a亚晶格上,Fe3+元素全部占据在16d亚晶格上,锌铁氧体的阳离子分布表达式为 (Zn2+)[Fe23+]O4,与MnFe2O4中阳离子占位分布变化相比,ZnFe2O4中的阳离子占位分布变化受温度影响较小,与现有文献中的实验测试结果和理论计算结果相吻合[33]。
表 2
ZnxMn1-xFe2O4体系中的阳离子占位情况
Table 2.
Site Occupation of ZnxMn1-xFe2O4
x(Zn) Temperature/K Tetrahedral Octahedral Method Resource Zn Mn Fe Zn Mn Fe 0.00 1 273 0.93 0.07 0.07 1.93 This work 1 273 0.93 0.07 0.07 1.93 XAFS [23] 1 473 0.91 0.09 0.09 1.91 This work 1 473 0.89 0.11 0.11 1.89 Mossbauer [24] 0.25 1 473 0.24 0.58 0.18 0.01 0.17 1.82 This work 0.50 1 573 0.50 0.25 0.25 0 0.25 1.75 This work 1 573 0.47 0.27 XRD [25] 0.75 1 473 0.75 0.02 0.23 0 0.23 1.77 This work 1 473 0.60 0.12 0.28 0.15 0.13 1.72 XRD [26] 1 473 0.63 0.16 0.21 0.12 0.09 1.79 XRD [26] 1.00 773 0.99 0.01 0.01 1.99 This work 773 0.98 0.02 0.02 1.98 XRD [27] 2.2 NixMn1-xFe2O4系中的阳离子分布随温度变化的规律
图 4为不同成分下NixMn1-xFe2O4体系中阳离子在8a和16d亚晶格上的占位分数随温度的变化趋势图,表 3为本文计算值与现有文献值的对照表。
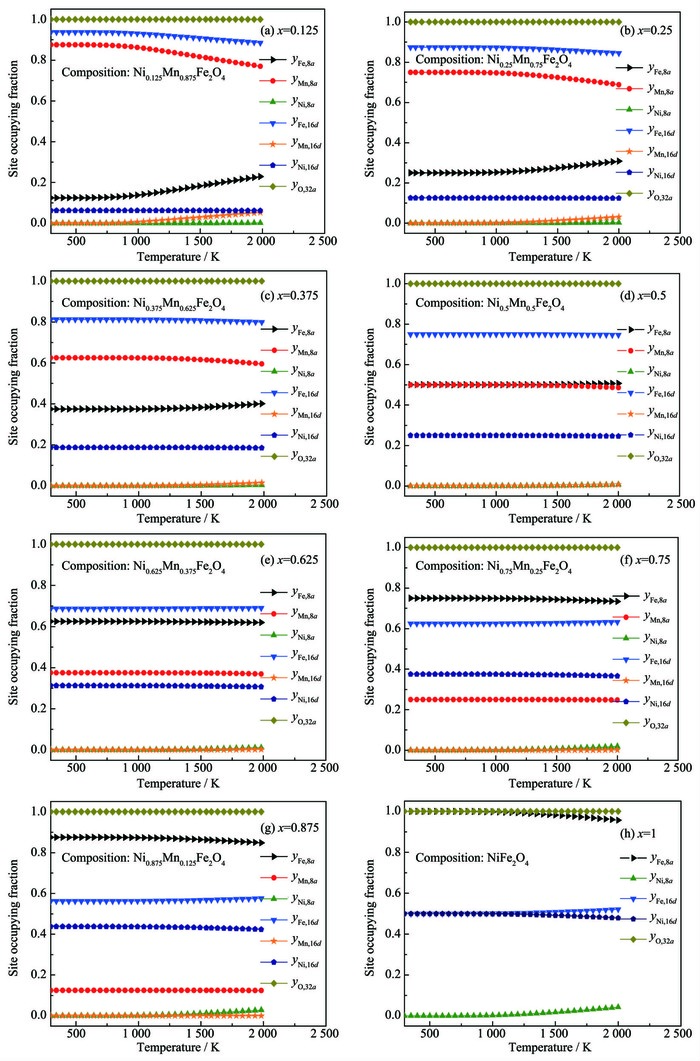 图 4
NixMn1-xFe2O4体系立方尖晶石中阳离子的占位分数随热处理温度和成分的变化趋势图
Figure 4.
Cation occupying fraction dependence on heat treatment temperature and composition in cubic spinel NixMn1-xFe2O4
表 3
NixMn1-xFe2O4体系中阳离子的占位结果
Table 3.
Site occupation of NixMn1-xFe2O4
图 4
NixMn1-xFe2O4体系立方尖晶石中阳离子的占位分数随热处理温度和成分的变化趋势图
Figure 4.
Cation occupying fraction dependence on heat treatment temperature and composition in cubic spinel NixMn1-xFe2O4
表 3
NixMn1-xFe2O4体系中阳离子的占位结果
Table 3.
Site occupation of NixMn1-xFe2O4
x(Ni) Temperature / K Tetrahedral 0ctahedral Method Resource Ni Mn Fe Ni Mn Fe 0 1 523 0.899 4 0.100 6 0.100 6 1.899 4 Calcd. This work 1 523 0 0.790 0 0.210 0 0 0.210 0 1.790 0 XRD [12] 1 523 0 0.764 9 0.235 1 0 0.235 1 1.7649 Calcd. [12] 0.125 1 523 0.000 2 0.813 8 0.185 9 0.124 8 0.061 2 1.8141 Calcd. This work 0.25 1 523 0.000 6 0.723 4 0.276 0 0.249 4 0.026 6 1.723 8 Calcd. This work 1 523 0.005 0 0.667 5 0.327 5 0.245 0 0.082 5 1.672 5 XRD [12] 1 523 0 0.654 6 0.345 4 0.250 0 0.095 4 1.6546 Calcd. [12] 0.375 1 523 0.001 2 0.6149 0.383 9 0.373 8 0.010 1 1.8141 Calcd. This work 0.5 1 523 0.002 2 0.496 5 0.501 2 0.497 8 0.003 5 1.498 8 Calcd. This work 1 523 0.0100 0.475 0 0.515 0 0.490 0 0.025 0 1.485 0 XRD [12] 1 523 0 0.475 0 0.525 0 0.500 0 0.025 0 1.475 0 Calcd. [12] 0.625 1 523 0.004 1 0.373 9 0.622 1 0.124 8 0.061 2 1.8141 Calcd. This work 0.75 1 523 0.007 0 0.249 7 0.743 4 0.743 0 0.000 3 1.256 6 Calcd. This work 1 523 0.002 0 0.242 5 0.750 7 0.748 0 0.002 5 1.249 5 XRD [12] 1 523 0 0.246 0 0.754 0 0.750 0 0.004 0 1.246 0 Calcd. [12] 0.875 1 523 0.011 6 0.1249 0.863 5 0.124 8 0.061 2 1.8141 Calcd. This work 1 1 523 0.0183 0 0.981 7 0.081 3 0 1.981 7 Calcd. This work 1 523 0 0 1 1 0 1 XRD [12] 1 523 0 0 1 1 0 1 Calcd. [12] 对比上述计算结果与对应文献结果发现,各个成分下的NixMn1-xFe2O4(x=0、0.25、0.5、0.75、1) 的占位结果均与文献结果符合得较好。特别对Ni2+的占位倾向预测结果与实验值相当吻合,证明了NiFe2O4是反式尖晶石结构。对Ni物质的量分数为1时,本文计算结果发现有微量的Ni2+会占据到8a亚晶格,这可能是因为在1 523 K下极少数的Ni2+与Fe3+互相交换位置,占位有序化减弱。但是,随着温度下降,由于Ni2+具有强烈占据16d亚晶格的倾向,其可能会随着温度降低而重新占据到16d亚晶格上。进一步分析Ni2+、Mn2+和Fe3+的变化规律发现,Ni2+强烈占据在16d亚晶格上,而Mn2+和Fe3+的占位有序化行为受到Ni2+的成分和温度的影响较大。当Ni2+的物质的量分数不大于0.5时,Mn2+和Fe3+的占位有序化行为受到温度影响较大,随温度升高,越来越多的Mn2+和Fe3+相互交换位置,占位有序化减弱;当Ni2+的物质的量分数大于0.5时,Mn2+其完全占据在8a亚晶格位置上,Fe3+完全占据在16d亚晶格位置,并且几乎不随温度的变化而发生变化。
通过对比已有NixMn1-xFe2O4(x=0、0.25、0.5、0.75、1) 中Ni2+、Mn2+以及Fe3+的占位有序化行为的文献结果与本文在相同成分时的计算结果,发现两者吻合得较好。证明了基于亚晶格模型,结合材料热力学与第一性原理计算的方法对预测镍锰铁氧体中阳离子占位有序化行为具有较高的可靠性。由于Wei等[12]已经对NixMn1-xFe2O4(x=0、0.25、0.5、0.75、1) 体系进行了系统研究,因此,本文研究了其他成分下NixMn1-xFe2O4(x=0.125、0.375、0.625、0.875) 中Ni2+、Mn2+以及Fe3+的占位有序化行为,并预测了各个成分在1 523 K时的占位结构。文献对这些成分的镍锰铁氧体研究的结果鲜有报道。因此,本文预测出的精细结构可以为相关材料设计提供重要的结构信息。
3 结论
本文基于热力学亚晶格模型,将材料热力学与第一性原理计算相结合,较为准确地预测了立方结构的尖晶石体系ZnxMn1-xFe2O4与NixMn1-xFe2O4中阳离子在亚晶格上的占位有序化行为随温度和成分的变化规律,得出结论如下:
(1) 在ZnxMn1-xFe2O4中,室温下Zn2+、Mn2+完全占据在8a亚晶格上,Fe3+完全占据在16d亚晶格上,属于正尖晶石结构,且Zn2+比Mn2+更倾向于占据在8a亚晶格上;随温度的升高,Zn2+与Mn2+均会与Fe3+发生位置交换行为,与实验结果吻合。本文也分析讨论了不同成分下的阳离占位有序化行为。
(2) 在NixMn1-xFe2O4中,Ni2+强烈地倾向于占据在16d亚晶格上,其占位几乎不随温度和成分的变化而发生变化,Mn2+倾向于占据在8a亚晶格上,而Fe3+倾向于占据在16d亚晶格上,仅在温度较高时发生部分位置交换,与实验结果吻合。
(3) 本文预测结果与可靠的文献结果吻合较好,表明采用热力学亚晶格模型,结合第一性原理计算和热力学计算的方法可以应用于铁氧体中阳离子的占位有序化行为预测,并且具有一定的普适性。
-
-
[1]
Taghavaei A H, Shokrollahi H, Janghorban K. J. Magn. Magn. Mater., 2009, 321:3926-3932 doi: 10.1016/j.jmmm.2009.07.061
-
[2]
Shokrollahi H. J. Magn. Magn. Mater., 2008, 320:463-474 doi: 10.1016/j.jmmm.2007.07.003
-
[3]
Wang Y M, Wang X, Jiang Y F, et al. Rare Met., 2006, 25: 531-535 doi: 10.1016/S1001-0521(07)60139-9
-
[4]
Sun Q C, Sims H, Mazumdar D, et al. Phys. Rev. B, 2012, 86:205106 doi: 10.1103/PhysRevB.86.205106
-
[5]
David S, Alberto R, Ricardo G, et al. Phys. Rev. B, 2015, 91: 195106 doi: 10.1103/PhysRevB.91.195106
-
[6]
Huang J R, Cheng C. J. Appl. Phys., 2013, 113:033912 doi: 10.1063/1.4776771
-
[7]
Zaag P J, Brabers V A M, Johnson M T, et al. Phys. Rev. B, 1995, 51:12009 doi: 10.1103/PhysRevB.51.12009
-
[8]
Mathur P, Thakur A, Singh M. Phys. Scr., 2008, 77:045701 doi: 10.1088/0031-8949/77/4/045701
-
[9]
Wang J, Zeng C, Peng Z M, et al. Physica B, 2004, 349:124-128 doi: 10.1016/j.physb.2004.02.014
-
[10]
Atsuto S, Koretaka Y, Fumiyasu O, et al. Phys. Rev. B, 2006, 73:184117 doi: 10.1103/PhysRevB.73.184117
-
[11]
Wei S H, Zhang S. Phys. Rev. B, 2001, 63:045112 doi: 10.1103/PhysRevB.63.045112
-
[12]
Wei Q M, Li J B, Chen Y J, et al. Mater. Chem. Phys., 2002, 74:340-343 doi: 10.1016/S0254-0584(01)00487-4
-
[13]
Sickafus K E, Wills J M, Grimes N W. J. Am. Ceram. Soc., 1999, 82:3279-3292
-
[14]
Mats H. J. Alloys Compd., 2001, 320:161-176 doi: 10.1016/S0925-8388(00)01481-X
-
[15]
Wu B, Zinkevich M, Aldinger F, et al. Intermetallics, 2008, 16:42-51 doi: 10.1016/j.intermet.2007.07.007
-
[16]
Anubhav J, Geoffroy H, Shyue P O, et al. Phys. Rev. B, 2011, 84:045115 doi: 10.1103/PhysRevB.84.045115
-
[17]
Stephan L. Phys. Rev. B, 2008, 78:245207 doi: 10.1103/PhysRevB.78.245207
-
[18]
ZHANG Chao-Hui(张朝辉), ZHANG Li-Kun(张立昆), WU Bo(吴波), et al.(中国稀土学报), 2011, 29:146-152
-
[19]
Wu B, Zinkevich M, Aldinger F, et al. J. Solid State Chem., 2007, 180:3280-3287 doi: 10.1016/j.jssc.2007.09.022
-
[20]
Kresse G, Hafner J. J. Phys.: Condens. Matter., 1994, 6:8245 doi: 10.1088/0953-8984/6/40/015
-
[21]
John P P, Kieron B, Matthias E. Phys. Rev. Lett., 1996, 77: 3865 doi: 10.1103/PhysRevLett.77.3865
-
[22]
Andersson J O, Thomas H, Lars H, et al. Calphad, 2002, 26: 273-312 doi: 10.1016/S0364-5916(02)00037-8
-
[23]
Yuki M, Hikaru K, Shinji K, et al. Surf. Interface Anal., 2010, 42:1655-1658 doi: 10.1002/sia.3621
-
[24]
Stefan P, Holger H, Marco S, et al. RSC Adv., 2013, 3:23001-23014 doi: 10.1039/c3ra44383c
-
[25]
Abbas T, Khan Y, Ahmad M, et al. Solid State Commun., 1992, 82:701-703 doi: 10.1016/0038-1098(92)90064-G
-
[26]
Attia S M. Egypt. J. Solids, 2006, 4:e340
-
[27]
O'neill H S C, Annersten H, Virgo D. Am. Mineral., 1992, 77:725-740
-
[28]
Szotek Z, Temmerman W, Ködderitzsch D, et al. Phys. Rev. B, 2006, 74:174431 doi: 10.1103/PhysRevB.74.174431
-
[29]
Henderson C M B, Charnock J M, Plant D A. J. Phys.: Condens. Matter., 2007, 19:076214 doi: 10.1088/0953-8984/19/7/076214
-
[30]
Fatemi D J, Harris V G, Browning V M, et al. J. Appl. Phys., 1998, 83:6867-6869 doi: 10.1063/1.367766
-
[31]
Dong H L, Hong S K, Chul H Y, et al. Mater. Chem. Phys., 1998, 57:169-172 doi: 10.1016/S0254-0584(98)00192-8
-
[32]
Kumara A M, Raju T R K P, Rao P A, et al. Int. J. Adv. Sci. Res. Sc. Technol., 2012, 1:1-4
-
[33]
Miller A. J. Appl. Phys., 1959, 30:S24-S25 doi: 10.1063/1.2185913
-
[1]
-

图 1 MnFe2O4的晶体结构
Figure 1 Crystal structure of MnFe2O4
Red balls are oxygen anions; Mn2+ in the purple tetrahedral interstices; Fe3+ in the yellow octahedral interstices

图 2 热力学函数计算路径转换示意图 (其中反应路径③ 可以转换为由路径① 和路径② 联合来实现,即③=①+②)
Figure 2 Alternative computing path of thermodynamic function denoting ③=①+②

图 3 ZnxMn1-xFe2O4体系立方尖晶石中阳离子的占位分数随热处理温度和成分的变化趋势图
Figure 3 Cation occupying fraction dependence on heat treatment temperature and composition in cubic spinel ZnxMn1-xFe2O4

图 4 NixMn1-xFe2O4体系立方尖晶石中阳离子的占位分数随热处理温度和成分的变化趋势图
Figure 4 Cation occupying fraction dependence on heat treatment temperature and composition in cubic spinel NixMn1-xFe2O4
表 1 立方相尖晶石结构铁氧体的晶体学结构信息
Table 1. Spinel (MgAl2O4-H11) Structure
Prototype Pearson's symbol Space Group Sublattice Atomic Coordinate MgAl2O4 cF56 Fd3m 227 Multiplicity Wyckoff notation Point symmetry x y z 8 a -43m 0 0 0 l6 d -3m 0.625 0.625 0.625 32 e 3m 0.375 0.375 0.375  下载: 导出CSV
下载: 导出CSV
表 2 ZnxMn1-xFe2O4体系中的阳离子占位情况
Table 2. Site Occupation of ZnxMn1-xFe2O4
x(Zn) Temperature/K Tetrahedral Octahedral Method Resource Zn Mn Fe Zn Mn Fe 0.00 1 273 0.93 0.07 0.07 1.93 This work 1 273 0.93 0.07 0.07 1.93 XAFS [23] 1 473 0.91 0.09 0.09 1.91 This work 1 473 0.89 0.11 0.11 1.89 Mossbauer [24] 0.25 1 473 0.24 0.58 0.18 0.01 0.17 1.82 This work 0.50 1 573 0.50 0.25 0.25 0 0.25 1.75 This work 1 573 0.47 0.27 XRD [25] 0.75 1 473 0.75 0.02 0.23 0 0.23 1.77 This work 1 473 0.60 0.12 0.28 0.15 0.13 1.72 XRD [26] 1 473 0.63 0.16 0.21 0.12 0.09 1.79 XRD [26] 1.00 773 0.99 0.01 0.01 1.99 This work 773 0.98 0.02 0.02 1.98 XRD [27]
下载: 导出CSV
表 3 NixMn1-xFe2O4体系中阳离子的占位结果
Table 3. Site occupation of NixMn1-xFe2O4
x(Ni) Temperature / K Tetrahedral 0ctahedral Method Resource Ni Mn Fe Ni Mn Fe 0 1 523 0.899 4 0.100 6 0.100 6 1.899 4 Calcd. This work 1 523 0 0.790 0 0.210 0 0 0.210 0 1.790 0 XRD [12] 1 523 0 0.764 9 0.235 1 0 0.235 1 1.7649 Calcd. [12] 0.125 1 523 0.000 2 0.813 8 0.185 9 0.124 8 0.061 2 1.8141 Calcd. This work 0.25 1 523 0.000 6 0.723 4 0.276 0 0.249 4 0.026 6 1.723 8 Calcd. This work 1 523 0.005 0 0.667 5 0.327 5 0.245 0 0.082 5 1.672 5 XRD [12] 1 523 0 0.654 6 0.345 4 0.250 0 0.095 4 1.6546 Calcd. [12] 0.375 1 523 0.001 2 0.6149 0.383 9 0.373 8 0.010 1 1.8141 Calcd. This work 0.5 1 523 0.002 2 0.496 5 0.501 2 0.497 8 0.003 5 1.498 8 Calcd. This work 1 523 0.0100 0.475 0 0.515 0 0.490 0 0.025 0 1.485 0 XRD [12] 1 523 0 0.475 0 0.525 0 0.500 0 0.025 0 1.475 0 Calcd. [12] 0.625 1 523 0.004 1 0.373 9 0.622 1 0.124 8 0.061 2 1.8141 Calcd. This work 0.75 1 523 0.007 0 0.249 7 0.743 4 0.743 0 0.000 3 1.256 6 Calcd. This work 1 523 0.002 0 0.242 5 0.750 7 0.748 0 0.002 5 1.249 5 XRD [12] 1 523 0 0.246 0 0.754 0 0.750 0 0.004 0 1.246 0 Calcd. [12] 0.875 1 523 0.011 6 0.1249 0.863 5 0.124 8 0.061 2 1.8141 Calcd. This work 1 1 523 0.0183 0 0.981 7 0.081 3 0 1.981 7 Calcd. This work 1 523 0 0 1 1 0 1 XRD [12] 1 523 0 0 1 1 0 1 Calcd. [12]
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 2254
- HTML全文浏览量: 514
 下载:
下载:
