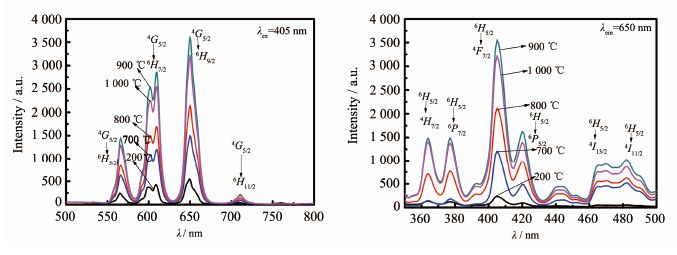
 图 6
Ca0.98Sm0.02WO4的发射和激发光谱图
Figure 6.
Emission spectra (λex405 nm) and excitation spectra (λem=650 nm) of Ca0.98Sm0.02WO4
图 6
Ca0.98Sm0.02WO4的发射和激发光谱图
Figure 6.
Emission spectra (λex405 nm) and excitation spectra (λem=650 nm) of Ca0.98Sm0.02WO4
引用本文:
吴锦绣, 李梅, 柳召刚, 胡艳宏, 王觅堂. 焙烧温度和掺杂浓度对Ca1-xSmxWO4发光性能的影响[J]. 无机化学学报,
2016, 32(1): 34-42.
doi:
10.11862/CJIC.2016.018

Citation: WU Jin-Xiu, LI Mei, LIU Zhao-Gang, HU Yan-Hong, WANG Mi-Tang. Effects of Doped Concentration and Calcination Temperature on Luminescence Properties of Ca1-xSmxWO4 Phosphors[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(1): 34-42. doi: 10.11862/CJIC.2016.018
Citation: WU Jin-Xiu, LI Mei, LIU Zhao-Gang, HU Yan-Hong, WANG Mi-Tang. Effects of Doped Concentration and Calcination Temperature on Luminescence Properties of Ca1-xSmxWO4 Phosphors[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(1): 34-42. doi: 10.11862/CJIC.2016.018
焙烧温度和掺杂浓度对Ca1-xSmxWO4发光性能的影响
摘要:
采用水热法制备了不同Sm3+掺杂量和不同焙烧温度的CaWO4:Sm3+系列荧光粉。用X射线衍射仪(XRD)、扫描电镜(SEM and EDS)、荧光分光光度计(FL)、傅里叶变换红外光谱仪(FIIR)和HORIBA Fluoromax-4仪等手段对样品的组成、结构、形貌、发光性质和量子效率进行分析和表征。分析结果表明:所得产物都为白钨矿结构。在405 nm近紫外光激发下,产物的发射光谱都有3个主发射峰组成,分别位于566、606和650 nm处,归属于Sm3+的4G5/2→6HJ/2(J=5,7,9)跃迁。随着Sm3+的物质的量分数的增加,样品发光强度先增强后减弱,当Sm3+的物质的量分数为1%时发光强度达到最高。对实验数据进行分析确定了钐离子间的能量传递类型为离子交换作用;并计算了能量传递的临界距离为2.46 nm。
-
关键词:
- CaWO4∶Sm3+
- / 水热法
- / 光致发光
English
Effects of Doped Concentration and Calcination Temperature on Luminescence Properties of Ca1-xSmxWO4 Phosphors
Abstract:
The Ca1-xSmxWO4 phosphors with different doping concentrations and different sintered temperatures were synthesized by hydrothermal method. The phase structure,morphology, luminescent properties and quantum efficiency of the as-synthesized samples were characterized by X-ray diffraction,fourier transform infrared spectroscopy, scanning electron microscope, fluorescence spectrophotometer respectively and Fluoromax-4. The results showed that all the phases were indexed to scheelite structure. The emission spectra of Ca1-xSmxWO4 phosphors exhibit three main peaks assigned to the 4G5/2→66HJ/2 (J=5,7,9) transitions of Sm3+ under 405 nm excited radiation, the dominating emission peaks at 566, 606, 650 nm. The luminescence intensity firstly increases with increasing of Sm3+ mole fraction, and then decreases. Experiments show that the best Sm3+ doping concentration is 1%. The energy transfer type between Sm3+ ions was determined to be the exchange interaction and the critical energy transfer distance (Dc) was calculated to be 2.46 nm.
-
Key words:
- CaWO4: Sm3+
- / hydrothermal method
- / photoluminescence
-
0 引言
钨酸盐是典型的自激活发光材料,其本征发光谱带很宽,几乎占据可见光区域的大部分,并且发光光谱十分稳定。钨酸盐可以由某些杂质激活,这些杂质被掺入钨酸盐点阵中之后,可使其具有特殊性质的发光。目前CaWO4已被广泛应用于医疗器械、X-射线增感屏、光纤设备、激光等领域[1-2]。稀土离子掺杂的发光材料也被广泛应用于固态激光器、光纤放大器、显示器、照明设备、生物传感器等领域[3-5],因此稀土掺杂的钨酸盐体系备受研究者们的关注。
在众多稀土离子中, 研究比较多的是三价Eu3+[6-8]。Eu3+掺杂CaWO4基质能够获得色纯度较高的红光,高杨等[9]和孟庆裕等[10]分别通过微乳液法和共沉淀法制备了Eu3+掺杂CaWO4红色荧光粉。刘运等[11]首次用水热法合成中空球形的CaWO4∶Eu3+。稀土Sm3+的4f-4f跃迁吸收峰位同样也位于近紫外区和蓝光区,并能够将吸收的能量转化为红光发射。Eu3+和Sm3+共掺杂的CaWO4红色荧光粉也有学者研究[12-13],该类荧光粉体系中引入Sm3+可以拓宽激发光谱至400 nm左右, 还可以增强其红光发射强度。孟庆裕等[14]用沉淀法制备了CaWO4∶Sm3+荧光粉,并研究其发光性质及其能量传递机理;Tian等[15] 用PVP作为模板剂,合成纺锤形的CaWO4∶Sm3+荧光粉,并研究其发光性能和机理。Yu等[16]用固相法合成新型橙红色Ba2CaWO4:Sm3+荧光粉。
不同的制备方法和条件会直接影响发光材料的微观形貌、颗粒尺寸和光致发光性能[17],从而影响其应用。大量研究表明,传统制备钨酸盐的高温固相法存在合成温度高、烧结时间长(1 100~1 200 ℃烧结数小时)、能耗大、荧光粉颗粒形貌差、粒度分布不均匀等缺点。近年来采用水热法制备无机纳米材料成为研究的热点。
本文用水热法制备了Sm3+掺杂的CaWO4材料前驱体,系统的研究了焙烧温度对产物的晶体结构和微观形貌的影响及其发光性能;重点研究了Sm3+含量对产物Ca1-xSmxWO4的光学性质的影响。分析了Sm3+的浓度猝灭机理和钐离子间的能量传递机理; 并计算了能量传递的临界距离为2.46 nm; 同时监测最佳产物的发光量子效率和色坐标。本课题的研究为开发利用稀土钐资源在发光领域的应用提供理论依据,同时为寻找性能优良的钨酸盐基发光材料具有一定的学术和应用意义。
1 实验部分
1.1 原料与试剂
CaCl2和Na2WO4(AR,天津市北联精细化学品开发有限公司),Sm2O3(纯度大于99.99%,包头稀土研究院),无水乙醇、浓硝酸和浓盐酸(AR,国药集团化学试剂有限公司),实验用水全为纯净水(自制)。
1.2 样品的制备
采用水热法合成Ca0.98Sm0.02WO4和Ca1-xSmxWO4(Sm3+的物质的量分数:0.6%、1.0%、2.0%、5.0%、8.0%)前驱体。按照合成需要的化学计量比称取一定量的CaCl2和Sm(NO3)3溶液混合均匀,然后缓慢逐滴加入到Na2WO4溶液中,产生白色沉淀,用4 mol·L-1 HCl或NaOH溶液调节pH值为7.5,在磁力加热搅拌器下搅拌10 min,然后转移到100 mL的高压反应釜的聚四氟乙烯中,填充度为80 mL,放入鼓风干燥箱中,设置水热温度为200 ℃、水热时间为36 h、阳离子与阴离子的物质的量配比为1∶2(水热条件是通过正交试验优化得到)。常温离心分离和洗涤得前驱体,然后将此前驱体置于刚玉坩埚内,放入马弗炉内,设置焙烧温度(700~1 000 ℃),在空气气氛中焙烧3 h,随炉冷却后得到最终产物。
1.3 样品的表征和性能测试
用德国的D8 ADVANCE X-射线粉末衍射仪进行物相分析,采用Cu Kα(λ=0.154 5 nm), 实施样品的晶型和纯度及其相的鉴定,设定收集角度为10°~80°, 扫描速度为4°·min-1;用荷兰飞利浦的QUANTA 400扫描电镜观测样品的晶体尺寸和形貌并确定产物的粒径大小和分布;并测定能谱确定产物的组成元素;采用日本的F-4600荧光分光光度计测定样品的激发和发射光谱,测试条件为激发和发射狭缝都为5 nm, 电压为400 V;傅里叶变换红外光谱仪是美国BROKER的ALPHAA,采用KBr压片法测定;产物的量子效率和色坐标用英国的HORIBA Fluoromax-4设备测试。所有样品都在室温下测试。
2 结果与讨论
2.1 焙烧温度的研究
2.1.4 激发和发射光谱分析
图 6为产物Ca0.98Sm0.02WO4在不同焙烧温度下的发射光谱(λex405 nm)和激发光谱图(λem=650 nm)。从图 6可知,产物在405 nm激发时,由于Sm3+中4f壳层电子之间f-f跃迁,都出现了4个荧光发射峰[19-22],位于566 nm处发射峰归属于4G5/2→6H5/2跃迁;位于606 nm处发射峰归属于4G5/2→6H7/2跃迁;位于650 nm处发射峰归属于4G5/2→6H9/2跃迁;位于700~710 nm处发射峰归属于4G5/2→6H11/2跃迁,这4个峰是Sm3+的特征发射峰。其中位于650 nm处的发射峰强度最大,且在606 nm处的发射峰都分裂为相邻两部分,这是由于产物的晶体场强度发生变化,从而引起Sm3+的能级发生劈裂的缘故。从图 6可以看出:不同焙烧温度的产物其发射峰的形状和位置基本一致,最强峰都位于650 nm处,其次是568 nm,因此产物为纯正红光。
图 6
Ca0.98Sm0.02WO4的发射和激发光谱图
Figure 6.
Emission spectra (λex405 nm) and excitation spectra (λem=650 nm) of Ca0.98Sm0.02WO4
前驱体的激发和发射光谱和焙烧后的最终产物的峰型和峰位基本一致,但强度都比较弱。随着焙烧温度的增加,产物的荧光强度逐渐增加,当焙烧温度达到900 ℃时,荧光强度达到最大,随着焙烧温度继续升高,产物的荧光强度呈下降趋势。这是由于在700 ℃时,多晶的固相反应还没有完全,只是部分形成了Sm3+掺杂的CaWO4,没有很好的形成发光中心,荧光强度较弱。当焙烧温度达到900 ℃时,Sm3+掺杂的CaWO4多晶形成的发光中心最多,因此荧光强度最大。当焙烧温度继续上升时,产物发光中心受到破坏,因而降低了发光强度。从图 6的激发光谱图(λem=650 nm)可以看出:从360到500 nm之间有多处激发光谱,即在363 nm(6H5/2→4H7/2)、377 nm(6H5/2→6P7/2)、405 nm(6H5/2→4F7/2)、420 nm(6H5/2→6P5/2)、440 nm(6H5/2→4G9/2)、465 nm(6H5/2→4I13/2)和482 nm(6H5/2→4I11/2)处,归属于Sm3+的内层电子间的f-f特征跃迁吸收[19-22],可知该类产物的激发光谱覆盖了 360~500 nm 很宽的区域。
从图 6可以看出:前驱体和不同焙烧温度的产物的激发峰的形状和位置基本一致,其最强吸收峰都位于405 nm,且900 ℃的产物激发峰最高。由以上分析可知:900 ℃焙烧所得产物的结晶度高、球形和类球形的形貌稳定和均匀、发光性能较好,所以制备Sm3+掺杂的CaWO4多晶的最佳焙烧温度为900 ℃。
2.1.3 红外光谱(IR)分析和能谱(EDS)
由图 4可以看出,焙烧前后的样品的振动类型非常相似,在750~850 cm-1之间都有一强烈的吸收带,是CaWO4中W-O键的伸缩振动峰。200 ℃的红外光谱图中在3 430~3 450 cm-1和1 630~1 640 cm-1的吸收峰是源于样品中残留或吸附溶剂水中的-OH的伸缩振动和弯曲振动[18-19]。而在900 ℃焙烧后的样品这2个峰就不明显,这是因为高温处理后,样品的表面比较纯净,吸附水被蒸发除去。为了进一步确定产物的元素组成,对样品进行能谱分析,如图 5所示,所制产物由O、W、Ca、Na和Sm 5种元素组成。可见红外光谱和能谱测定进一步证实产物为纯相的白钨矿,与XRD分析的结果相一致。
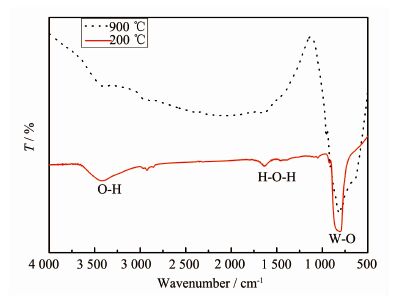 图 4
Ca0.98Sm0.02WO4的焙烧前(200 ℃)和焙烧后(900 ℃)的红外光谱图
Figure 4.
FTIR spectra of Ca0.98Sm0.02WO4 samples before sintering (200 ℃) and after sintering (900 ℃)
图 4
Ca0.98Sm0.02WO4的焙烧前(200 ℃)和焙烧后(900 ℃)的红外光谱图
Figure 4.
FTIR spectra of Ca0.98Sm0.02WO4 samples before sintering (200 ℃) and after sintering (900 ℃)
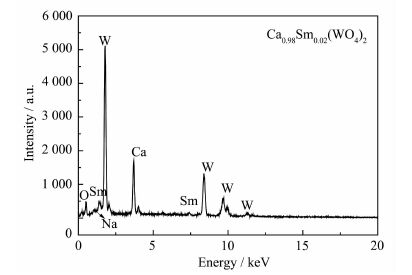 图 5
Ca0.98Sm0.02WO焙烧后(900 ℃)能谱图
Figure 5.
EDS of Ca0.98Sm0.02WO4 after sintering (900 ℃)
图 5
Ca0.98Sm0.02WO焙烧后(900 ℃)能谱图
Figure 5.
EDS of Ca0.98Sm0.02WO4 after sintering (900 ℃)
2.1.1 产物的结构分析
图 1为前驱体(200 ℃水热合成)和分别在700,800,900,1 000 ℃焙烧得到最终产物的XRD图。其X射线衍射峰位置与体心四方晶系CaWO4标准卡片(PDF#41-1431 )XRD图完全一致,标准卡片的晶胞参数为a=b=52.4 nm, c=113.7 nm。产物没有出现其它峰,说明无论是前驱体还是最终产物都是纯相白钨矿结构。在产物晶格中1个Sm3+取代1个Ca2+产生了电荷不平衡。由于原材料选用Na2WO4,所以反应体系中有大量的Na+, 因而由1个Sm3+和1个Na+取代2个Ca2+使产物晶格电荷平衡,这是个自发过程。虽然掺杂了2%的Sm3+,但对CaWO4晶体结构并未引起变化,只是峰位向大角度略微偏移,说明Sm3+和Na+没有进入到晶格间隙中,而是以替代Ca2+的形式存在于晶格结构中。这是由于Sm3+的物质的量比较少,且Sm3+(0.096 nm)和Na+(0.102 nm)离子半径与Ca2+(0.098 nm)的离子半径相近,容易占据Ca2+的格位进入CaWO4基质中,故对CaWO4的晶体结构几乎没有影响。图中200 ℃为水热合成的前驱体,其衍射峰比较宽,说明产物的颗粒尺寸接近微纳米颗粒。随着焙烧温度的增加,产物的衍射峰宽度变窄,强度升高。焙烧温度为900 ℃时,在(112)、(200)、(204)、(116)和(312)这5个主要特征衍射峰达到最强,这说明焙烧温度900 ℃的产物结晶性能最好,当温度进一步升高时,由图中曲线1 000 ℃可知,(004)和(213)晶面增强,而其它衍射峰的强度却减少,这可能是由于焙烧温度过高容易使晶体长大,甚至部分产物的晶格将发生畸变。
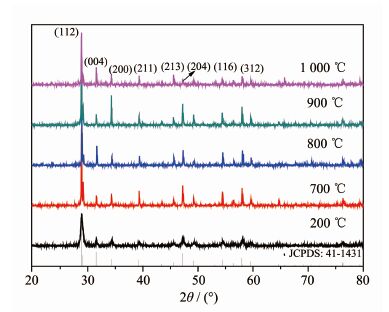 图 1
Ca0.98Sm0.02WO4在不同焙烧温度下的XRD图
Figure 1.
XRD patterns of Ca0.98Sm0.02WO4 at different sintered temperatures
图 1
Ca0.98Sm0.02WO4在不同焙烧温度下的XRD图
Figure 1.
XRD patterns of Ca0.98Sm0.02WO4 at different sintered temperatures
2.1.2 产物微观形貌的分析
从图 2可知:水热法合成的前驱体颗粒为球形,表面比较疏松,粒径为1 μm左右,颗粒分散均匀。通过700 ℃焙烧后,产物为不规则多边形,表面比较致密,粒径为0.5~1 μm,颗粒分散不均匀,出现团聚现象。这可能是由于产物在700 ℃焙烧时,产物变致密,从而使该温度焙烧后的产物的粒径比焙烧前小。从图 3可知,随着焙烧温度的升高,产物颗粒由不规则向球形过渡,粒径随着温度的逐渐升高而逐渐变大,且分散均匀,基本没有发生团聚。800 ℃焙烧后,颗粒仍为不规则多边形,粒径长到3~4 μm;900 ℃焙烧后,颗粒为类球形,粒径长到5~6 μm;1 000 ℃焙烧后,产物颗粒基本为球形和椭球形,粒径基本变化不大,仍为5~6 μm。这可能是由于在焙烧过程中,随着温度的升高,粒径逐渐长大,并且表面越来越致密和圆滑,到900 ℃,产物的粒径和形貌变化不大,趋于稳定状态。 这与XRD分析的结果相吻合。从图 2和图 3可知,产物的形貌基本都为球形和类球形,可见产物的形貌具有遗传性。
 图 2
Ca0.98Sm0.02WO4在焙烧前(200 ℃)和焙烧后(700 ℃)的扫描电镜图
Figure 2.
SEM image of Ca0.98Sm0.02WO4 at Sintering before(200 ℃) and after sintering (700 ℃)
图 2
Ca0.98Sm0.02WO4在焙烧前(200 ℃)和焙烧后(700 ℃)的扫描电镜图
Figure 2.
SEM image of Ca0.98Sm0.02WO4 at Sintering before(200 ℃) and after sintering (700 ℃)
 图 3
Ca0.98Sm0.02WO4在不同温度焙烧后的扫描电镜图
Figure 3.
SEM image of Ca0.98Sm0.02WO4 at different sintered temperatures
图 3
Ca0.98Sm0.02WO4在不同温度焙烧后的扫描电镜图
Figure 3.
SEM image of Ca0.98Sm0.02WO4 at different sintered temperatures
3.2 Sm3+含量的分析
3.2.2 激发和发射光谱
图 9为Ca1-xSmxWO4(三价钐离子的物质的量分数为0.6%、1.0%、2.0%、5.0%和8.0%,水热反应后,焙烧900 ℃)在650 nm监测下的激发光谱图,由图 9可知,掺杂不同Sm3+浓度的钨酸钙,其发射峰的形状和位置基本一样,都由两部分组成,其一是在200~ 280 nm处所有样品均呈现一个较高的宽峰,其最高峰位(249 nm)因掺杂浓度的差异而有所不同,是属于O2-→W6+和O2-→Sm3+电荷迁移带叠加而成[10, 19, 21];其二是从320 nm到500 nm有7个窄带,这个范围内的线状激发峰来自于Sm3+的4f-4f跃迁,从左到右依次为在363 nm(6H5/2→4H7/2)、377 nm(6H5/2→6P7/2)、405 nm(6H5/2→4F7/2)、420 nm(6H5/2→6P5/2)、440 nm(6H5/2→4G9/2)、465 nm(6H5/2→4I13/2)及其482 nm(6H5/2→4I11/2)跃迁,最强的4f-4f激发峰位于405 nm处。由图 9所示最强激发峰是Sm3+物质的量分数为1%时的样品。
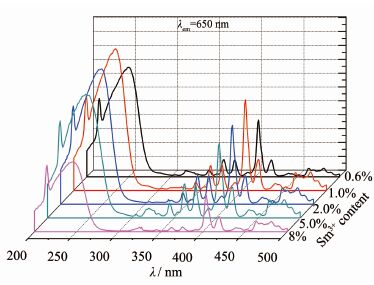 图 9
Ca1-xSmxWO4在λem=650 nm的激发光谱图
Figure 9.
Excitation spectra (λem=650 nm) of Ca1-xSmxWO4
图 9
Ca1-xSmxWO4在λem=650 nm的激发光谱图
Figure 9.
Excitation spectra (λem=650 nm) of Ca1-xSmxWO4
图 10为Ca1-xSmxWO4在249和405 nm激发下的发射光谱图。从图 10中的λex249 nm发射光谱图可看出,在350~550 nm有一个宽带发射峰,为基质CaWO4的自激发发射峰,即W6+→O2电荷迁移态的发射。观察所有发射谱可以看到, 随着Sm3+掺杂浓度的增加,W6+→O2电荷迁移态的发光强度在逐渐减弱, 而与图 9的基质激发峰在200~280 nm的变化规律不一致,这主要是由于在CaWO4∶Sm3+样品中WO42-将能量传递给Sm3+,从而使Sm3+的发射强度增强, 而WO42-自身的发射强度降低。在500~700 nm之间是Sm3+的4f-4f跃迁特征发射峰,分别位于 566、606和 650 nm处,分别归属于Sm3+的4G5/2→ 66HJ/2(J=5,7,9)跃迁。随着Sm3+的物质的量分数的增加,样品发光强度先增强后减弱,当Sm3+的物质的量分数为1%时发光强度达到最高,说明在Sm3+含量为1%的产物中,基质的能量向Sm3+传递的效率最高。图 10中λex405 nm激发的发射峰,同样都显示3个主要的发射峰,其中位于650 nm处发射峰强度最大;且在606 nm处的发射峰都分裂为相邻两部分;Sm3+的物质的量分数为1%时的样品发光强度达到最高。由以上分析可知Sm3+掺杂浓度为1%时荧光强度最强。当掺杂物质的量分数高于1%时,随着Sm3+浓度增加荧光强度逐渐减弱,发生了浓度猝灭现象。这是由于掺杂高浓度Sm3+时,Sm3+之间的距离拉近,彼此之间有能量传递,降低了发光强度。在低浓度范围内增加Sm3+的浓度,Sm3+提供的发光中心增多,因此荧光强度随Sm3+浓度增强而增强。可见在本论文所研究的体系中Sm3+猝灭浓度为1%,即Sm3+的最佳掺杂物质的量分数为1%。
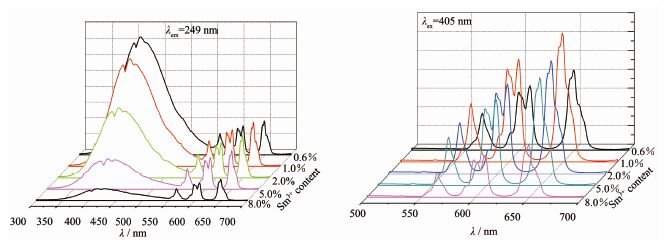 图 10
Ca1-xSmxWO4在λex249 nm和λex405 nm的发射光谱图
Figure 10.
Emission spectra (λex249 nm and λex405 nm) of Ca1-xSmxWO4
图 10
Ca1-xSmxWO4在λex249 nm和λex405 nm的发射光谱图
Figure 10.
Emission spectra (λex249 nm and λex405 nm) of Ca1-xSmxWO4
3.2.1 晶体结构和形貌
图 7为Sm3+掺杂物质的量分数为1%和8%的样品在900 ℃焙烧的XRD图。从图中可以看出,制备出的样品的X射线衍射峰位置与体心四方晶系CaWO4标准卡片(PDF#41-1431)XRD图完全一致,没有出现其它杂峰,说明产物为纯相的CaWO4,Sm3+和Na+都取代Ca2+进入晶格中。但Sm3+掺杂浓度为1%的衍射峰比8%的样品强,这可能是Sm3+掺杂浓度为1%的样品结晶性能比较好的原因。从图 8的扫描电镜照片中,我们看到产物形貌都为类球形,Sm3+掺杂浓度为1%的粒径为5 μm左右,而Sm3+掺杂物质的量分数为8%的产物颗粒不均匀,粒径为5~10 μm左右,这可能是由于钐含量太多,晶格生长不完全而导致的。
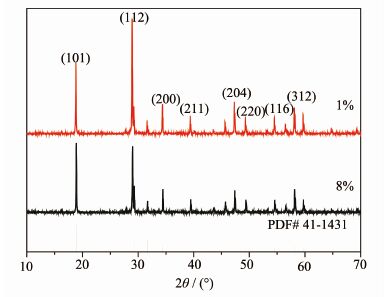 图 7
在900 ℃焙烧Ca1-xSmxWO4样品的XRD图
Figure 7.
XRD patterns of Ca1-xSm xWO4 samples synthetised in 900 ℃
图 7
在900 ℃焙烧Ca1-xSmxWO4样品的XRD图
Figure 7.
XRD patterns of Ca1-xSm xWO4 samples synthetised in 900 ℃
 图 8
Ca1-xSmxWO4的扫描电镜图(Sm3+的物质的量分数为1%和8%)
Figure 8.
SEM patterns of Ca1-xSmxWO4 (Sm3+ molar percentage 1% and 8%)
图 8
Ca1-xSmxWO4的扫描电镜图(Sm3+的物质的量分数为1%和8%)
Figure 8.
SEM patterns of Ca1-xSmxWO4 (Sm3+ molar percentage 1% and 8%)
3.3 浓度猝灭机理和能量传递临界距离的探讨
能量传递是稀土离子之间相当普遍的一种现象, 主要是通过离子间的能级匹配来进行能量交换的物理过程。因为稀土离子有丰富的能级, 尤其是在晶体中, 由于晶体场的作用能级发生了劈裂, 能级匹配的机会也随之增多, 就会出现稀土离子间的能量传递和浓度猝灭等现象。根据Dexter理论[24-25],非导电性无机材料中激活剂离子的浓度猝灭机理属于电多极相互作用,发光强度I与激活剂浓度x之间遵循如下关系:
I/x∝(βxs/3)-1或lg (I/x)=C-(s/3)lgx
式中x为激活剂浓度,I为样品的发光强度,β是常数,s为电多级相互作用的指数。以lgx为横坐标,lg(I/x)为纵坐标做图,得到一条直线,斜率为s/3。若当s=3时,代表离子间的交换作用;当s=6时,代表电偶极-电偶极相互作用;s=8为电偶极-电四极相互作用;s=10为电四极-电四极相互作用[26]。
图 11为Sm3+掺杂浓度大于最佳浓度(x=1.0%)时lg(I/x)与lgx的关系图, I为发射峰(566、606和653 nm)强度。利用Origin7.5软件对图 10中的实验点进行线性拟合,得出3条平行线,3个发射峰的lg(I/x)与lgx基本都呈线性关系(相关系数分别为0.982 9、0.985 5、0.983 3), 斜率分别为-1.356、-1.357、-1.356, 基本都接近1,由此斜率可求出 s值为3,因此在CaWO4基质中Sm3+中心的浓度猝灭主要是钐离子间交换作用引起的。
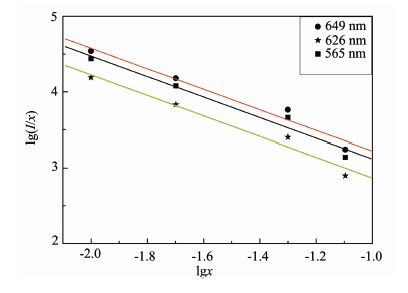 图 11
产物Ca1-xSmxWO4的lg(I/x)与lgx的关系
Figure 11.
Relationship Between lg(I/x) and lgx of Ca1-xSmxWO4
图 11
产物Ca1-xSmxWO4的lg(I/x)与lgx的关系
Figure 11.
Relationship Between lg(I/x) and lgx of Ca1-xSmxWO4
能量传递的临界距离是指浓度猝灭发生时发光中心之间的平均距离。稀土离子间的能量传递主要有电多极相互作用和交换相互作用两大类型。稀土离子能量传递的临界距离可以表示为[10]:
上式中V是一个晶胞的体积,N是一个晶胞中可被替位的阳离子的数目,C是临界(猝灭)浓度。对于四方相CaWO4晶体,N=4,V=0.312 64 nm3,Sm3+的猝灭浓度C=0.01。代入公式中可以得出Dc=2.46 nm, 可见能量传递的临界距离为2.46 nm。
3.4 量子效率和色坐标的研究
发光材料辐射出的量子数与吸收的激发量子数之比称为量子效率。一种好的荧光粉应具备充分吸收激发态能量且尽可能高效转化为发射光的特点。换而言之,荧光粉的量子效率应最大化。本文最佳产物Ca0.99Sm0.01WO4焙烧前的量子效率为4.57,色坐标x=0.571 58,y=0.427 8。该样品经过900 ℃焙烧后量子效率提高到18.24,色坐标为x=0.592 69,y=0.406 58。说明焙烧有利于该系列的荧光粉提高量子效率和色纯度。
4 结论
本文采用水热法合成Ca1-xSmxWO4前驱体,然后研究焙烧温度对发光性能的影响;最后研究钐离子的物质的量分数对产物发光性能的影响。用XRD、扫描电镜、荧光光谱仪和傅里叶变换红外光谱仪等手段对样品的组成、结构、形貌及其发光性质进行分析和表征。详细的讨论相结构、形貌和Sm3+掺杂浓度对材料发光性能的影响和规律。XRD分析结果表明:产物晶型都为体心四方晶系的白钨矿结构。扫描电镜表明:随着焙烧温度升高,产物的颗粒尺寸逐渐增大,到900 ℃颗粒尺寸比较稳定,直径为5~6 μm的类球形形貌。荧光光谱仪分析表明样品在λex405 nm激发下,都出现3个主要的发射峰,分别位于566 nm(4G5/2→6H5/2)、606 nm(4G5/2→6H7/2)、650 nm(4G5/2→6H9/2)。Sm3+的最佳掺杂物质的量分数为1%,当Sm3+掺杂物质的量分数大于1%,出现浓度猝灭现象,主要是由钐离子间的交换作用引起的,能量传递的临界距离为2.46 nm。最佳产物Ca0.99Sm0.01WO4经过900 ℃焙烧后量子效率为18.24, 色坐标为x=0.592 69,y=0.406 58,产物为红色发光。
-
-
[1]
耿秀娟,田彦文,陈永杰.材料导报:综述篇, 2010,24(7):54-57GENG Xiu-Juan, TIAN Yan-Wen, CHEN Yong-Jie, et al. J. Mater. Rev. A, 2010,24(7):54-57
-
[2]
Lou X M, Chen D H. Mater. Lett., 2008,62:1681-1684 doi: 10.1016/j.matlet.2007.09.066
-
[3]
Ryu J H, Bang S Y, Kim W S, et al. J. Alloys Compd., 2007, 441:146-151 doi: 10.1016/j.jallcom.2006.07.130
-
[4]
冯晓辉,孟庆裕,孙江亭.物理学报, 2011,60(3):037806(9 pages)FENG Xiao-Hui, MENG Qin-Yu, SUN Jiang-Ting, et al. Acta Phys. Sin., 2011,60(3):037806(9 pages)
-
[5]
徐叙熔,苏勉曾.发光学与发光材料,2004.XU Xu-Rong, SU Mian-Zeng. Luminescence and Luminescent Materials, Beijing: Chemical Industry Press, 2004.
-
[6]
刘艳,姜营营,刘桂霞.无机化学学报, 2013,29(2):277-282 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130209&flag=1LIU Yan, JIANG Ying-Ying, LIU Gui-Xia , et al. Chinese J. Inorg. Chem., 2013,29(2):277-282 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130209&flag=1
-
[7]
周贤菊,陈加,杨小东.无机化学学报, 2012,28 (5):932-936 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20120512&flag=1ZHOU Xian-Ju, CHEN Jia, YANG Xiao-Dong . Chinese J. Inorg. Chem., 2012,28 (5):932-936 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20120512&flag=1
-
[8]
Li H Y, Noh H M, Moon B K, et al. Inorg. Chem., 2013,52: 11210-11217 doi: 10.1021/ic401453e
-
[9]
孟庆裕,张庆,李明.物理学报, 2012,61(10):107804 (8 Pages)MENG Qin-Yu, ZHANG Qi, LI Ming, et al. Acta Phys. Sin., 2012,61(10):107804 (8 Pages)
-
[10]
高杨,吕强,汪洋.物理学报, 2012,61(7):077802(9Pages)GAO Yang, LV Qiang ,WANG Yang, et al. Acta Phys. Sin., 2012,61(7):077802(9Pages)
-
[11]
刘运,温时瑜,李晋阳.发光学报, 2014,35(10): 1201-1204 doi: 10.3788/fgxbLIU Yun, WEN Shi-Yu, LI Jin-Yang , et al. Chinese J. Lumin., 2014,35(10): 1201-1204 doi: 10.3788/fgxb
-
[12]
Kang F G, Hun Y H, Wu H Y, et al. J. Lumin., 2012,132: 887-894 doi: 10.1016/j.jlumin.2011.11.022
-
[13]
Dabre K V, Dhoble S J. J. Lumin., 2014,150:55-58 doi: 10.1016/j.jlumin.2014.01.045
-
[14]
毕长虹,孟庆裕.物理学报, 2013,62(19):197801(7 Pages)BI Chang-Hong, MENG Qin-Yu. Acta Phys. Sin. , 2013,62(19):197801(7 Pages)
-
[15]
Tian Y, Liu Y, Hua R N, et al. Mater. Res. Bull., 2012,47: 59-62 doi: 10.1016/j.materresbull.2011.10.007
-
[16]
Ruiijn Y, Hyeon M N, Byung K M, et al. J. Lumin., 2014, 152:133-137 doi: 10.1016/j.jlumin.2014.01.074
-
[17]
翟永清,李漩,李金航.人工晶体学报, 2014,43(5): 1061-1066ZHAI Yong-Qing, LI Xuan, LI Jin-Hang , et al. J. Synth. Cryst., 2014,43(5): 1061-1066
-
[18]
Wang W X, Yang P P, Gai Sh L, et al. J. Nanopart. Res., 2010,12(6):2295-2305 doi: 10.1007/s11051-010-9850-4
-
[19]
Maheshwary B P, Singh R A. New J. Chem., 2015,39:4494 -4507 doi: 10.1039/C4NJ01911C
-
[20]
Liu X, Qiao X S, Fan X P. Solid State Sci., 2011,13(3):579 -583 doi: 10.1016/j.solidstatesciences.2010.12.029
-
[21]
吴锦绣,李梅,柳召刚.无机化学学报, 2015,31(3):452-458 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20150304&flag=1WU Jin-Xiu, LI Mei, LIU Zhang-Gang , et al. Chinese J. Inorg. Chem., 2015,31(3):452-458 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20150304&flag=1
-
[22]
Yang Z P, Dong H Y, Liu P F, et al. J. Rare Earths, 2014,32(5):404-408 doi: 10.1016/S1002-0721(14)60085-5
-
[23]
Mo F W, Zhou L Y, Pang Q, et al. J. Ceram. Int., 2012(38): 6289-6294
-
[24]
Dexter D I, Jnmes H S. Chem. Phys., 1954,22(6):1063-1070
-
[25]
关丽,魏伟,刘超.硅酸盐学报, 2013,41(1):62-65GUAN Lin, WEI Wei, LIU Chao, et al. J. Chin. Ceram. Soc., 2013,41(1):62-65
-
[26]
Du H Y, Sun J F, Xia Z G. J. Electrochem. Soc., 2009,156 (12):J361-J366 doi: 10.1149/1.3236634
-
[1]
-

图 1 Ca0.98Sm0.02WO4在不同焙烧温度下的XRD图
Figure 1 XRD patterns of Ca0.98Sm0.02WO4 at different sintered temperatures

图 2 Ca0.98Sm0.02WO4在焙烧前(200 ℃)和焙烧后(700 ℃)的扫描电镜图
Figure 2 SEM image of Ca0.98Sm0.02WO4 at Sintering before(200 ℃) and after sintering (700 ℃)

图 3 Ca0.98Sm0.02WO4在不同温度焙烧后的扫描电镜图
Figure 3 SEM image of Ca0.98Sm0.02WO4 at different sintered temperatures

图 4 Ca0.98Sm0.02WO4的焙烧前(200 ℃)和焙烧后(900 ℃)的红外光谱图
Figure 4 FTIR spectra of Ca0.98Sm0.02WO4 samples before sintering (200 ℃) and after sintering (900 ℃)

图 5 Ca0.98Sm0.02WO焙烧后(900 ℃)能谱图
Figure 5 EDS of Ca0.98Sm0.02WO4 after sintering (900 ℃)

图 6 Ca0.98Sm0.02WO4的发射和激发光谱图
Figure 6 Emission spectra (λex405 nm) and excitation spectra (λem=650 nm) of Ca0.98Sm0.02WO4

图 7 在900 ℃焙烧Ca1-xSmxWO4样品的XRD图
Figure 7 XRD patterns of Ca1-xSm xWO4 samples synthetised in 900 ℃

图 8 Ca1-xSmxWO4的扫描电镜图(Sm3+的物质的量分数为1%和8%)
Figure 8 SEM patterns of Ca1-xSmxWO4 (Sm3+ molar percentage 1% and 8%)

图 9 Ca1-xSmxWO4在λem=650 nm的激发光谱图
Figure 9 Excitation spectra (λem=650 nm) of Ca1-xSmxWO4

图 10 Ca1-xSmxWO4在λex249 nm和λex405 nm的发射光谱图
Figure 10 Emission spectra (λex249 nm and λex405 nm) of Ca1-xSmxWO4
-


 下载:
下载:










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1100
- HTML全文浏览量: 127
 下载:
下载:
