 图 1
Fe3O4 (a)、Fe3O4@C (b)、Fe3O4@C@LDH (c) 和Fe3O4@LDO (d) 的XRD图 (●针铁矿)
Figure 1.
XRD patterns of Fe3O4 (a), Fe3O4@C (b), Fe3O4@C@LDH (c) and Fe3O4@LDO (d) (● goethite phase)
图 1
Fe3O4 (a)、Fe3O4@C (b)、Fe3O4@C@LDH (c) 和Fe3O4@LDO (d) 的XRD图 (●针铁矿)
Figure 1.
XRD patterns of Fe3O4 (a), Fe3O4@C (b), Fe3O4@C@LDH (c) and Fe3O4@LDO (d) (● goethite phase)
引用本文:
盘登科. 空心结构磁性固体碱催化剂Fe3O4@LDO的制备与性能[J]. 无机化学学报,
2016, 32(1): 74-80.
doi:
10.11862/CJIC.2016.007

Citation: PAN Deng-Ke. Synthesis and Properties of the Hollow Magnetic Solid Based Catalyst Fe3O4@LDO[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(1): 74-80. doi: 10.11862/CJIC.2016.007
Citation: PAN Deng-Ke. Synthesis and Properties of the Hollow Magnetic Solid Based Catalyst Fe3O4@LDO[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(1): 74-80. doi: 10.11862/CJIC.2016.007
空心结构磁性固体碱催化剂Fe3O4@LDO的制备与性能
摘要:
基于水滑石类化合物的复合氧化物 (LDO) 是一类性能优异的固体碱催化剂,对其进行改性和功能化引起了越来越多的关注。本文将空心结构和Fe3O4引入到镁铝复合氧化物中,制备了一种空心结构磁性固体碱催化剂Fe3O4@LDO。这种空心结构磁性固体碱催化剂粒子具有以镁铝复合氧化物为壳层,空心Fe3O4为核的核壳结构。由于其独特的空心结构,Fe3O4@LDO粒子的悬浊液具有良好的稳定性,将其应用于催化Knoevenagel缩合反应,达到平衡后苯甲醛的转化率约为62%,显示出较好的催化性能。同时,Fe3O4@LDO粒子具有较强的磁性,非常方便分离与回收,是一种性能优良的磁性固体碱催化剂。
English
Synthesis and Properties of the Hollow Magnetic Solid Based Catalyst Fe3O4@LDO
Abstract:
The mixed oxides based on layered double hydroxide (LDO) are novel solid base catalyst, and their modification and functionalization have received more and more interesting in the area of catalysis. In this paper, the hollow magnetic solid base catalyst has been (Fe3O4@LDO) synthesized by coating LDO onto the hollow Fe3O4 particles. The Fe3O4@LDO particles possess well defined core-shell structure with Mg-Al Mixed Oxides as the shell and hollow Fe3O4 particle as the core. Owing to its unique hollow structure, the Fe3O4@LDO particles possess high colloid stability, and showing promising application in homogeneous catalysis. In the Knoevenagel condensation of benzaldehyde with ethyl acetoacetate under solvent free conditions, the Fe3O4@LDO particles gave the highest activity with yield of 62% in 2 h, showing high catalytic activity. In the mean time, the Fe3O4@LDO particles have strong magnetic response, can be easily separated and recycled after a catalyst reaction.
-
Key words:
- hollow magnetic particles
- / core shell structure
- / Fe3O4
- / LDO
- / solid base catalyst
-
在一定温度下,通过焙烧水滑石类化合物 (LDH) 得到的复合金属氧化物 (LDO) 具有丰富的碱中心和孔道结构,以及较大的比表面积,是一种性能优良的固体碱催化剂和催化剂载体[1-4]。但人们发现LDO催化剂的分离和回收很难实现,极大的影响了其在催化领域里的应用[5-8],对其进行改性和功能化的需求日益迫切。近年来,将磁性物质引入LDO的研究引起了人们的广泛关注。如Zhang等采用共沉淀法将镁铝尖晶石引入到镁铝水滑石中并焙烧成磁性复合氧化物,发现其在丙酮自缩合反应中具有较好的催化性能[5]。Li等[7]采用层层组装法将剥离后的水滑石层板包覆在经二氧化硅修饰的四氧化三铁表面制备了一种磁性复合氧化物,负载W7O246-后进行了六氯化苯的光降解实验,发现其是性能优异的磁性催化剂载体。
在前期工作中[8],我们制备了一种具有核壳结构和较强磁性的磁性镁铝复合氧化物粒子,并对包覆层镁铝复合氧化物的厚度进行了有效的调控。但在制备过程,我们发现所制备的前体Fe3O4@C磁性粒子的胶体或悬浮液的稳定性较差,尽管预先采用长达20 min的超声分散处理,依然有许多磁性粒子没有包覆上目的物,而是以沉淀的形式与产物分离,所得目的产物也出现了包覆层不均匀的现象,需要进行二次包覆才能得到较好形态的磁性复合氧化物。显然,这些缺点不利于这类磁性复合氧化物在催化领域特别是均相催化中的应用。
研究发现,将磁性粒子制备成空心结构可极大的降低其密度,从而提高其胶体或悬浊液的稳定性[9-12]。因此,将空心结构引入Fe3O4@C磁性粒子并进一步制备磁性固体碱催化剂是解决上述问题的一种行之有效的方法。本研究首先采用溶剂热法合成了一种空心Fe3O4@C磁性粒子,然后采用共沉淀法将硝酸根插层LDH包覆到空心Fe3O4@C磁性粒子的表面,之后经500 ℃焙烧2 h得到空心磁性复合氧化物Fe3O4@LDO。采用XRD、IR、SEM、TEM和VSM等表征了Fe3O4@LDO的晶体结构、组成、形貌以及磁学性能。另外,将所制备的Fe3O4@LDO用于催化苯甲醛和氰乙酸乙酯的Knoevengel缩合反应,发现其具有较好的催化性能,并且非常容易分离和回收,是一种性能优良的磁性固体碱催化剂。
1 实验部分
1.1 试剂与仪器
FeCl3·6H2O和CH3COONa·3H2O为分析纯 (北京益利精细化学品有限公司),葡萄糖,硝酸和乙二醇均为分析纯,无水乙醇为化学纯 (北京化工厂),脱二氧化碳去离子水实验室自制。
日本岛津XRD-6000型X射线粉末衍射仪测定样品的晶体结构 (40 kV,30 mA,Cu Kα射线,波长0.154 18 nm,扫描速度为5°·min-1(2θ),防散射狭缝为1°,扫描范围3°~70°,接受狭缝为0.15 mm)。Vector-22型傅立叶红外光谱仪,利用KBr压片法测定在4 000~400 cm-1的红外光谱图。采用日本Shimadzu公司ICPS-7500型元素分析仪对样品的Mg、Al、Fe元素含量进行分析 (载气流量0.7 L·min-1, 冷却气流量14 L·min-1,等离子气流量1.2 L·min-1)。采用JDM-13型振动样品磁强计,室温下测试,最大磁场15 kOe,称量样品20 mg,测试产物的比饱和磁化强度和矫顽力。Hitachi S-4700扫描电子显微镜 (SEM) 用于分析样品表面形貌。用Hitachi-800透射电子显微镜表征样品的核壳结构,适量样品分散于乙醇水溶液中 (50%乙醇,体积比),超声振荡处理20 min后滴于铜网上, 溶剂自然挥发后进行TEM分析。岛津GC-MS 2010型气质联用仪分析催化反应产物的组成。
1.2 制备
1.3 空心Fe3O4@LDO磁性粒子的催化性能研究
以Knoevenagel缩合为探针反应研究了空心Fe3O4@LDO磁性粒子的催化性能[15-16]。将苯甲醛 (50 mmol) 和氰基乙酸乙酯 (50 mmol) 加入50 mL的三口瓶中,加入50 mg Fe3O4@LDO,N2保护下,磁力搅拌,60 ℃下反应。每隔一定时间取样1 μL,用5 mL丙酮稀释,采用岛津GC-MS 2010型气质联用仪分析产物组成。采用相同方法对LDO的催化性能也进行了研究。
1.2.1 空心Fe3O4@C磁性粒子的制备
采用溶剂热法制备Fe3O4磁性粒子[13]。具体如下:称取2.7 g (0.01 mol) FeCl3·6H2O,7.2 g (0.045 mol) CH3COONa·3H2O,溶解于400 mL乙二醇中,室温下搅拌至形成分散均匀的黄色溶液。将所得到的溶液加入500 mL高压釜中,然后在200 ℃下晶化8 h。将高压釜取出水浴冷却至室温,将所得到的黑色悬浊液用无水乙醇和去离子水各洗涤3次,洗涤时采用永磁铁分离出黑色固体,得到粒径大小为100 nm左右的磁性粒子Fe3O4。称取0.2 g Fe3O4,加入150 mL 0.1 mol·L-1的HNO3,超声10 min使Fe3O4凝胶化,然后磁性分离,再用去离子水洗涤1次,磁性分离出Fe3O4。另称取葡萄糖9.91 g (0.05 mol),完全溶解于150 mL去离子水中,加入Fe3O4后混合超声20 min,将所得到悬浊液加入200 mL的高压釜中,在180 ℃下碳化反应4 h。所得产品洗涤过程与Fe3O4相同,然后保存在无水乙醇中并命名为Fe3O4@C。
1.2.2 空心Fe3O4@LDO磁性粒子的制备
利用Fe3O4@C中碳膜上丰富的功能基团和水滑石粒子间的相互作用力[8, 14],将硝酸根插层水滑石包覆在Fe3O4@C粒子表面,然后焙烧得到空心Fe3O4@LDO磁性粒子。具体操作如下:量取含有0.12 g Fe3O4@C的悬浊液,磁性分离。称取0.42 g NaOH与Fe3O4@C一并加入60 mL甲醇中,超声至NaOH完全溶解得到溶液A。另称取0.769 2 g (0.003 mol) Mg (NO3)2·6H2O和0.563 0 g (0.001 5 mol) Al (NO3)2·9H2O,加入60 mL甲醇,超声至固体完全溶解得到溶液B。在N2保护条件下将B快速滴入A中,调整溶液pH值为9.5,65 ℃搅拌并晶化48 h,得到黑色悬浊液,磁性分离,用脱CO2去离子水洗涤。所得到的黑色固体分散到甲醇中保存,命名为Fe3O4@C@LDH。将所得Fe3O4@C@LDH真空干燥,研磨后装入焙烧管,预先通N2 30 min,在N2保护条件下以1 ℃·min-1升温至500 ℃,恒温2 h,然后冷却至室温,得焙烧样品记为Fe3O4@LDO。元素分析结果表明Fe3O4@LDO中LDO的质量含量约为33.4%(由Mg、Al和Fe元素的比值换算得到,LDO视为MgO和Al2O3的混合物)。为了对比,采用相同方法制备了由硝酸根插层LDH焙烧而得的镁铝复合氧化物LDO。
2 结果与讨论
2.1 结构与组成
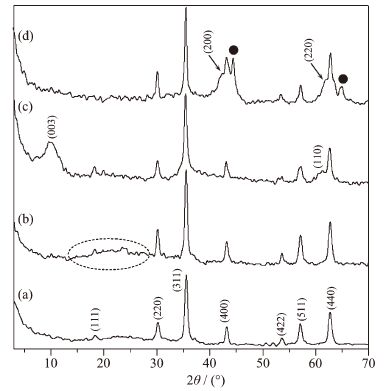
图 1a、b是Fe3O4和Fe3O4@C的XRD图。由图中可以看出,所制备的Fe3O4的XRD图中出现了典型的 (111)、(220)、(311)、(400)、(422)、(511) 和 (440) 等归属于Fe3O4的特征晶面衍射峰,与标准卡片 (PDF No.19-0629) 一致,说明制备了晶形规整的Fe3O4。当在Fe3O4表面包覆上一层碳膜后,Fe3O4@C (图 1b) 中不仅出现了归属于Fe3O4的特征衍射峰,在2θ角为15°~30°之间 (图 1b中虚线圈区域) 还出现了一系列归属于碳的特征衍射峰[8, 17]。由于生成的碳层为无定形碳,其衍射峰并不明显,与文献报道一致[8]。可以看出,除了衍射强度稍有变强外,Fe3O4@C中归属于Fe3O4的衍射峰没有发生变化,表明其晶体结构在包覆过程中并没有改变。Fe3O4的衍射峰变强可能是由于在包覆碳层过程中,Fe3O4粒子进一步熟化所致。图 1c、d是Fe3O4@C@LDH和Fe3O4@LDO的XRD图。Fe3O4@C@LDH (图 1c) 中不仅出现了归属于Fe3O4的特征衍射峰,还出现了归属于LDH的特征衍射峰 (003) 和 (110),表明所制备的产品是一种由LDH相和Fe3O4相组成的复合物。其中LDH相中 (003) 峰形宽化,可能是由于包覆水滑石过程中CO2进入反应体系,同时包覆了硝酸根插层LDH和碳酸根插层LDH所致[8]。相对于纯相的Fe3O4和LDH,Fe3O4@C@LDH中Fe3O4和LDH的衍射峰均较弱。LDH的衍射峰变弱可能是由于Fe3O4对其晶体生长具有抑制作用所致[8, 18]。而Fe3O4的衍射峰变弱则可能是形成了特殊的包覆结构,Fe3O4的衍射峰部分被覆盖所致。经500 ℃焙烧2 h后 (图 1d),Fe3O4@LDO中LDH的特征衍射峰消失,出现了微弱的归属于MgO的 (200) 和 (220) 衍射峰[8, 19],且归属于Fe3O4的特征衍射峰依然存在,表明所制备的Fe3O4@LDO主要成分是Fe3O4和镁铝复合氧化物。同时,图中出现了归属于针铁矿 (标识为●) 的衍射峰[20-21],这是由于在高温下,部分Fe3O4发生了晶相转变,生成了针铁矿所致。
图 1
Fe3O4 (a)、Fe3O4@C (b)、Fe3O4@C@LDH (c) 和Fe3O4@LDO (d) 的XRD图 (●针铁矿)
Figure 1.
XRD patterns of Fe3O4 (a), Fe3O4@C (b), Fe3O4@C@LDH (c) and Fe3O4@LDO (d) (● goethite phase)
图 2是所制备样品的FT-IR谱图。Fe3O4的FT-IR谱图 (图 2a) 中3 445 cm-1处对应于样品中羟基的伸缩振动吸收。在588 cm-1处出现了对应于Fe-O的伸缩振动吸收峰[8, 22],说明合成了Fe3O4。图 2b是Fe3O4@C的FT-IR谱图,谱图中1 617 cm-1是Fe3O4@C中C=C键的振动吸收,而1 411 cm-1是Fe3O4@C中Fe3O4表面吸附的CO32-的红外吸收,1 000~1 300 cm-1之间的吸收峰对应于样品中C-OH的振动吸收[8, 22-23]。588 cm-1处的吸收峰对应于Fe-O振动吸收,但是其强度较Fe3O4中弱,这可能是由于Fe3O4表面包覆上一层碳膜所致。这说明在Fe3O4@C中形成了以Fe3O4为核,碳为壳层的包覆结构。图 2c、d是Fe3O4@C@LDH和Fe3O4@LDO的FT-IR谱图。由图可知,Fe3O4@C@LDH的红外光谱中出现了1 383 cm-1吸收峰,归属于硝酸根的红外振动吸收。1 360 cm-1处微弱的吸收峰对应于碳酸根的红外吸收,可能与其含量较小有关[8, 22-23]。588 cm-1处出现了Fe-O的吸收峰,表明所制备的产品是一种由硝酸根LDH相和Fe3O4相组成的复合物。经过500 ℃焙烧2 h后 (图 2d),1 383 cm-1和1 360 cm-1处的吸收峰消失,而在623 cm-1处出现了归属于镁铝氧化物的红外吸收峰[8, 22, 24],表明形成了镁铝复合氧化物。另外,588 cm-1处Fe-O的吸收峰依然存在,表明产品中存在Fe3O4。以上表征结果表明经过焙烧后得到的产物是镁铝复合氧化物与Fe3O4形成的一种磁性复合氧化物。
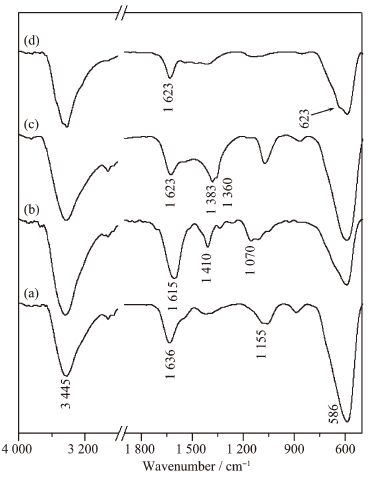 图 2
Fe3O4 (a)、Fe3O4@C (b)、Fe3O4@C@LDH (c) 和Fe3O4 @LDO (d) 的红外吸收谱图
Figure 2.
FT-IR spectra of Fe3O4 (a), Fe3O4@C (b), Fe3O4@C@LDH (c) and Fe3O4@LDO (d)
图 2
Fe3O4 (a)、Fe3O4@C (b)、Fe3O4@C@LDH (c) 和Fe3O4 @LDO (d) 的红外吸收谱图
Figure 2.
FT-IR spectra of Fe3O4 (a), Fe3O4@C (b), Fe3O4@C@LDH (c) and Fe3O4@LDO (d)
2.2 空心结构
图 3是Fe3O4和Fe3O4@C的SEM和TEM图。由图 3a可知,Fe3O4具有规整的球形结构,粒径约为160 nm,粒径分布均匀,分散性良好。仔细观察发现Fe3O4纳米粒子表面比较粗糙,由许多个10 nm左右的小粒子团聚而成的,与文献报道的Fe3O4类似[8, 13, 25]。但本研究中所制备的Fe3O4粒子具有更致密的堆砌结构,可能是其一次粒子粒径较小所致。图 3b是Fe3O4@C的SEM图片。相对于Fe3O4粒子的SEM图片,Fe3O4@C粒子表面变得相对光滑,与文献中所制备的碳球类似[26],表明在Fe3O4的表面包覆上了一层碳膜[8, 23, 25]。在包覆完成后Fe3O4依然保持了完好的球形形貌,其粒径约为240 nm,并具有典型的核壳结构。如图 3d所示,Fe3O4@C中碳包覆层厚度约为30 nm,其Fe3O4核大小约为150~200 nm,远大于其前体Fe3O4。另外,Fe3O4@C中Fe3O4核的形貌也与其前体Fe3O4有着显著差别。Fe3O4具有致密的堆砌结构,而Fe3O4@C中Fe3O4核则显示出较为松散多孔的形貌;同时,Fe3O4@C中的Fe3O4核具有十分典型的环形结构,与文献报道中的空心球十分相似[9-12, 27],表明成功制备了具有空心结构的Fe3O4@C粒子。图 3b插图是偶尔观察到的破损的单个Fe3O4@C粒子,可以明显的看出其具有中空结构,进一步证实了所制备的Fe3O4@C粒子具有空心结构。必须指出的是,Fe3O4@C粒子同时具有核壳结构和空心结构,而且致密连续的碳膜包覆在空心Fe3O4球表面,提供了丰富的如羧基、羟基等功能基团,可使LDH能更容易、更好的包覆在Fe3O4表面;空心Fe3O4核则使其具有相对较低的密度,可以显著增强其胶体或悬浮液的稳定性。
 图 3
Fe3O4 (a) 和Fe3O4@C (b) 的扫描电镜图; Fe3O4 (c) 和Fe3O4@C (d) 的透射电镜图
Figure 3.
SEM images of Fe3O4 (a) and Fe3O4@C (b); TEM images of Fe3O4 (c) and Fe3O4@C (d)
图 3
Fe3O4 (a) 和Fe3O4@C (b) 的扫描电镜图; Fe3O4 (c) 和Fe3O4@C (d) 的透射电镜图
Figure 3.
SEM images of Fe3O4 (a) and Fe3O4@C (b); TEM images of Fe3O4 (c) and Fe3O4@C (d)
另外,对Fe3O4@C空心粒子的形成机理也进行了初步的探讨。仔细观察可以发现,组成Fe3O4@C核层的Fe3O4一次粒子粒径约为20 nm左右,远大于其前体Fe3O4的一次粒子 (10 nm左右)。采用Scherrer公式[18]计算得到Fe3O4沿 (311) 和 (440) 晶面方向上的粒径D311和D440分别为7.6 nm和7.4 nm。包覆一层碳膜后,Fe3O4@C中Fe3O4的D311和D440显著增大,分别为23.3 nm和22.5 nm。以上结果表明在包覆过程中,Fe3O4不但形成了空心结构,而且存在着Fe3O4晶体增长过程。显然,Fe3O4晶体增长对空心Fe3O4@C粒子的形成具有至关重要的作用。据此,我们尝试给出空心Fe3O4@C粒子的形成机理。首先,经由溶剂热法制得的Fe3O4粒子必须具有较小粒径的一次粒子。在包覆过程中,碳纳米粒子在Fe3O4粒子表面沉积,并形成一层碳膜。与此同时,Fe3O4一次粒子发生Oswald熟化过程[11-12, 28],处于Fe3O4核内部的一次粒子逐渐溶解,并向外部迁移,留下许多孔隙,使Fe3O4核形成了空心结构,而处于Fe3O4核外部的一次粒子由于晶体增长,其粒径增大,进而形成了如图 3d中的空心粒子。值得一提的是,表面包覆的碳膜使Fe3O4粒子的晶体增长在一个相对封闭的环境中进行,避免了Fe3O4粒子在形成空心结构过程发生破裂,利于其后续应用。
图 4a是Fe3O4@C@LDH的扫描电镜图片。由图可知,Fe3O4@C@LDH的粒径分布比较均匀,粒子大小约为260 nm。与Fe3O4@C的光滑表面形貌不同,Fe3O4@C@LDH表面为相互交联堆积的LDH粒子,并且LDH粒子大小和形状不规则,只有极少数粒子出现了片状结构,晶型较差,这与LDH粒子较小而且粒子间相互作用较强有关[14, 18],与XRD表征结果一致。图 4c是Fe3O4@C@LDH的TEM图片。可以看出,Fe3O4@C@LDH粒子整体粒径相对于Fe3O4@C变大,并具有较清晰的核壳结构。壳层厚度约为40 nm,由一层形状不规则的LDH构成。由于表面水滑石层的遮挡,空心结构没有Fe3O4@C明显,但还是可以看出其具有明显的孔洞结构。将Fe3O4@C@LDH在500 ℃下焙烧2 h后,得到Fe3O4@LDO,如图 4b所示。Fe3O4@LDO显示出完全不同于Fe3O4@C@LDH的形貌,仅有的水滑石的片状结构消失,形成了相对光滑、致密堆积的小球,这与文献所报道的LDH转变为镁铝复合氧化物的形貌变化是一致的[8, 25]。由于碳层和壳层水滑石的层间阴离子被烧除,Fe3O4@LDO的粒径相对于Fe3O4@C@LDH的粒径有所变小,约为200 nm。图 4d是Fe3O4@LDO的TEM图片,可以看出,Fe3O4@ LDO粒径均匀,分散性较好。由于壳层和核层均为氧化物,Fe3O4@LDO的核壳结构并不十分明显,依稀可以看出壳层厚度约为25 nm,而其空心结构相对于Fe3O4@C@LDH却更明显了,可能是由于水滑石层和碳层被烧除所致。结合XRD和FT-IR表征结果可知,采用水滑石前体法成功制备了具有核壳结构的空心磁性固体碱催化剂。
 图 4
Fe3O4@C@LDH (a) 和Fe3O4@LDO (b) 的扫描电镜图; Fe3O4@C@LDH (c) Fe3O4@LDO (d) 的透射电镜图
Figure 4.
SEM images of Fe3O4@C@LDH (a) and Fe3O4@LDO (b); TEM images of Fe3O4@C@LDH (c) and Fe3O4 @LDO (d)
图 4
Fe3O4@C@LDH (a) 和Fe3O4@LDO (b) 的扫描电镜图; Fe3O4@C@LDH (c) Fe3O4@LDO (d) 的透射电镜图
Figure 4.
SEM images of Fe3O4@C@LDH (a) and Fe3O4@LDO (b); TEM images of Fe3O4@C@LDH (c) and Fe3O4 @LDO (d)
2.3 磁性
图 5是空心磁性固体碱催化剂Fe3O4@LDO及其前体的磁滞回线图。由图可知,Fe3O4的比饱和磁化强度为73.5 emu·g-1,具有较强的磁性。当包覆上一层碳膜,并形成空心结构后,Fe3O4@C的比饱和磁化强度稍有降低,为55.6 emu·g-1。包覆上硝酸根插层水滑石后,Fe3O4@C@LDH的比饱和磁化强度有较大幅度降低,约为40.5 emu·g-1,归因于Fe3O4含量的降低。焙烧后得到的Fe3O4@LDO的磁性为26.7 emu·g-1,依然具有较强的磁性。对空心磁性固体碱催化剂Fe3O4@LDO粒子的磁响应性能和重分散性能也进行了研究,结果如图 5插图所示。可以看出,Fe3O4@LDO粒子在水溶液中具有很好的分散效果,而将其置于一个0.2 T的永磁体附近时,Fe3O4@LDO粒子在20 s内就被吸引到靠近磁体的一侧,这表明Fe3O4@LDO粒子具有很好的磁响应性能。移除永磁铁并经过轻微振荡后,发现Fe3O4 @LDO粒子又能重新分散在溶液中,表明其具有很好的重分散性能,非常适用于均相催化反应中。
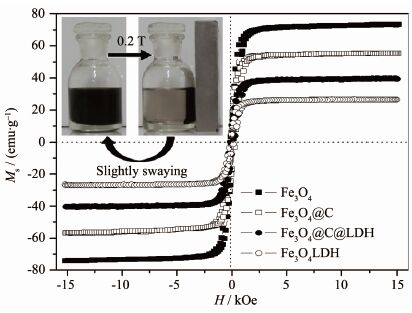 图 5
Fe3O4、Fe3O4@C、Fe3O4@C@LDH和Fe3O4@LDO的磁滞回线图
Figure 5.
Room temperature magnetization curves of Fe3O4, Fe3O4@C, Fe3O4@C@LDH and Fe3O4@LDO
图 5
Fe3O4、Fe3O4@C、Fe3O4@C@LDH和Fe3O4@LDO的磁滞回线图
Figure 5.
Room temperature magnetization curves of Fe3O4, Fe3O4@C, Fe3O4@C@LDH and Fe3O4@LDO
2.4 催化性能研究
以上表征结果表明,所制备的空心磁性固体碱催化剂Fe3O4@LDO磁性粒子不仅具有较强的磁性和规整的核壳结构,还具有空心结构,提升了其在溶液中的稳定性,非常适合作为催化剂,特别是在均相催化反应中。我们以苯甲醛和氰基乙酸乙酯为反应物,研究了空心Fe3O4@LDO粒子在Knoevenagel缩合反应中的催化性能[15-16]。为了对比,对采用相同方法制备的LDO粒子的催化性能也进行了研究。图 6是Fe3O4@LDO催化Knoevenagel缩合反应中苯甲醛的转化率随时间的变化关系图。由图可知,采用Fe3O4@LDO作为催化剂时,反应120 min后达到平衡,苯甲醛的转化率为62%。采用相同质量的纯相LDO作为催化剂时,苯甲醛的转化率为64%。由此可见,引入Fe3O4和空心结构后,Fe3O4@LDO粒子与纯相LDO的催化活性相差不大,表现出较好的催化性能。Fe3O4@LDO粒子较纯相LDO催化活性物质含量更少的情况下,达到了几乎相同的催化效果,一方面可能是由于LDO的催化性能较好,很少的催化剂用量就能达到很好的催化效果;另一方面,可能是由于LDO均匀地包覆在空心Fe3O4粒子表面,其分散性能更好,与反应物接触面积增大,从而使LDO的催化性能有所提升。另外,Fe3O4@LDO粒子具有较强的磁性,分离和回收再利用非常方便,是一种性能优良的磁性固体碱催化剂。
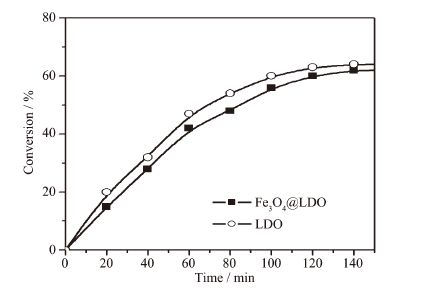 图 6
Fe3O4@LDO催化Knoevenagel缩合反应的产率随时间的变化关系
Figure 6.
Catalytic activity of Fe3O4@LDO in Knoevenagel condensation reaction: conversion of benzaldehyde as a function of reaction time
图 6
Fe3O4@LDO催化Knoevenagel缩合反应的产率随时间的变化关系
Figure 6.
Catalytic activity of Fe3O4@LDO in Knoevenagel condensation reaction: conversion of benzaldehyde as a function of reaction time
3 结论
采用溶剂热法合成了具有空心结构的Fe3O4@C磁性粒子,然后将镁铝硝酸根插层LDH包覆到Fe3O4@C粒子表面,在一定条件下焙烧后,成功制备了粒径均匀的空心磁性固体碱催化剂Fe3O4@LDO粒子。空心磁性固体碱催化剂Fe3O4@LDO粒子具有以镁铝复合氧化物为壳层,空心Fe3O4为核的核壳结构,其比饱和磁化强度为26.7 emu·g-1,具有较强的磁性。由于其独特的空心结构,Fe3O4@LDO粒子的胶体或悬浊液的稳定性较好,非常适用于均相催化反应。将空心磁性固体碱催化剂Fe3O4@LDO粒子用于催化苯甲醛和氰乙酸乙酯的Knoevengel缩合反应,发现其具有较好的催化性能,并且非常容易分离和回收,是一种性能优良的磁性固体碱催化剂。
-
-
[1]
Fan G, Li F, Evans D G, et al. Chem. Soc. Rev., 2014, 43: 7040-7066 doi: 10.1039/C4CS00160E
-
[2]
Lü W, Yang L, Fan B, et al. Chem. Eng. J., 2015, 263:309-316 doi: 10.1016/j.cej.2014.11.009
-
[3]
Zhang Z, Liao M, Zeng H, et al. Fuel Process. Technol., 2014, 128:519-524 doi: 10.1016/j.fuproc.2014.08.015
-
[4]
Jyothi T M, Raja T, Rao B S. J. Mol. Catal. A: Chem., 2001, 168:187-191 doi: 10.1016/S1381-1169(00)00527-6
-
[5]
Zhang H, Qi R, Evans D G, et al. J. Solid State Chem., 2004, 177:772-780 doi: 10.1016/j.jssc.2003.09.009
-
[6]
张慧, 徐彦红.化学学报, 2004, 62:750-756ZHANG Hui, XU Yan-Hong, EVANS D G, et al. Acta Chim. Sinica, 2004, 62:750-756
-
[7]
Li L, Feng Y, Li Y, et al. Angew. Chem. Int. Ed., 2009, 48: 5888-5892 doi: 10.1002/anie.v48:32
-
[8]
盘登科, 张慧.无机化学学报, 2011, 7:1341-1347 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20110720&flag=1PANG Deng-Ke, ZHANG Hui. Chinese J. Inorg. Chem., 2011, 7:1341-1347 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20110720&flag=1
-
[9]
Hu J, Chen M, Fang X, et al. Chem. Soc. Rev., 2011, 40: 5472-5491 doi: 10.1039/c1cs15103g
-
[10]
Luo B, Xu S, Ma W, et al. J. Mater. Chem., 2010, 20:7107-7113 doi: 10.1039/c0jm00726a
-
[11]
Sun Q, Ren Z, Wang R, et al. J. Nanopart. Res., 2011, 13: 213-220 doi: 10.1007/s11051-010-0020-5
-
[12]
Chen Y, Xia H, Lu L, et al. J. Mater. Chem., 2012, 22:5006-5012 doi: 10.1039/c2jm15440d
-
[13]
Deng H, Li X, Peng Q, et al. Angew. Chem. Int. Ed., 2005, 44:2782-2785 doi: 10.1002/(ISSN)1521-3773
-
[14]
Gursky J A, Blough S D, Luna C, et al. J. Am. Chem. Soc., 2006, 128:8376-8377 doi: 10.1021/ja0612100
-
[15]
Ebitani K, Motokura K, Mori K, et al. J. Org. Chem., 2006, 71(15):5440-5447 doi: 10.1021/jo060345l
-
[16]
郤玉生. 北京化工大学硕士学位论文. 2010.XI Yu-Sheng. Thesis for the Master of Beijing University of Chemical Technology. 2010.
-
[17]
Kong L, Lu X, Bian X, et al. ACS Appl. Mater. Interfaces, 2011, 3:35-42 doi: 10.1021/am101077a
-
[18]
Zhang H, Pan D, Zou K, et al. J. Mater. Chem., 2009, 19: 3069-3077 doi: 10.1039/b820176e
-
[19]
Wang Z L, Wang E B, Gao L, et al. J. Solid State Chem., 2005, 178:736-741 doi: 10.1016/j.jssc.2004.11.005
-
[20]
Gong C, Chen D, Jiao X, et al. J. Mater. Chem., 2002, 12: 1844-1847 doi: 10.1039/b201243j
-
[21]
Liang X, Wang X, Zhuang J, et al. Adv. Funct. Mater., 2006, 16:1805-1813 doi: 10.1002/(ISSN)1616-3028
-
[22]
Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. 5th Ed. New York: John Wiley and Sons, 1997.
-
[23]
Yin Y, Zhou S, Chen M, et al. J. Colloid Interface Sci., 2011, 361:527-533 doi: 10.1016/j.jcis.2011.05.014
-
[24]
Gunawan P, Xu R. Chem. Mater., 2009, 21:781-783 doi: 10.1021/cm803203x
-
[25]
Pan D, Zhang H, Fan T, et al. Chem. Commun., 2011, 47: 908-910 doi: 10.1039/C0CC01313G
-
[26]
孙晓明. 清华大学博士学位论文. 2005.SUN Xiao-Ming. Thesis for the Doctorate of Tsinghua University. 2005.
-
[27]
Wang Z, Wu L, Chen M, et al. J. Am. Chem. Soc., 2009, 131:11276-11277 doi: 10.1021/ja903246e
-
[28]
Liu S, Xing R, Lu F, et al. J. Phys. Chem. C, 2009, 113: 21042-21047 doi: 10.1021/jp907296n
-
[1]
-

图 1 Fe3O4 (a)、Fe3O4@C (b)、Fe3O4@C@LDH (c) 和Fe3O4@LDO (d) 的XRD图 (●针铁矿)
Figure 1 XRD patterns of Fe3O4 (a), Fe3O4@C (b), Fe3O4@C@LDH (c) and Fe3O4@LDO (d) (● goethite phase)

图 2 Fe3O4 (a)、Fe3O4@C (b)、Fe3O4@C@LDH (c) 和Fe3O4 @LDO (d) 的红外吸收谱图
Figure 2 FT-IR spectra of Fe3O4 (a), Fe3O4@C (b), Fe3O4@C@LDH (c) and Fe3O4@LDO (d)

图 3 Fe3O4 (a) 和Fe3O4@C (b) 的扫描电镜图; Fe3O4 (c) 和Fe3O4@C (d) 的透射电镜图
Figure 3 SEM images of Fe3O4 (a) and Fe3O4@C (b); TEM images of Fe3O4 (c) and Fe3O4@C (d)
Inset in (b): a single cracked Fe3O4@C particle

图 4 Fe3O4@C@LDH (a) 和Fe3O4@LDO (b) 的扫描电镜图; Fe3O4@C@LDH (c) Fe3O4@LDO (d) 的透射电镜图
Figure 4 SEM images of Fe3O4@C@LDH (a) and Fe3O4@LDO (b); TEM images of Fe3O4@C@LDH (c) and Fe3O4 @LDO (d)

图 5 Fe3O4、Fe3O4@C、Fe3O4@C@LDH和Fe3O4@LDO的磁滞回线图
Figure 5 Room temperature magnetization curves of Fe3O4, Fe3O4@C, Fe3O4@C@LDH and Fe3O4@LDO
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1324
- HTML全文浏览量: 169
 下载:
下载:
