
Scheme 1.
Enantioselective addition of thiols to ortho-quinone methides (o-QMs).

After ground-breaking works devoted by Akiyama [1], Terada and Uraguchi [2], last decade has seen exponential growth in the field of chiral phosphoric acids [3]. This family has become one of the most powerful organic catalysts. New generations of chiral phosphoric acids were continuously developed to catalyze a broad range of reactions with good to perfect enantioselectivities. In a common sense, a chiral phosphoric acid acts as a bifunctional Brønsted acid/base catalyst, in which the nonbonding electron pair of the oxygen atom in the P=O bond is regarded as a basic moiety and the P—OH hydrogen as an acidic moiety. However, in some cases, chiral phosphoric acids just solely act as Brønsted acid catalysts. This review is organized by the type of reactions catalyzed by chiral phosphoric acids.
Quinone methides (e.g., o-QMs and p-QMs) are highly active organic intermediates which can easily react with nucleophiles to give Michael addition type products. Chiral phosphoric acid catalysts, which bind the QMs (generated in situ by CPAs) and the nucleophiles together through hydrogen bonds, can stereoselectively catalyze 1, 4 or 1, 6-conjugate additions to afford enantio-enriched products from racemic starting materials.
Based on their early exploration works on the asymmetric 1, 4 and 1, 6-conjugate addition of carbon- [4] and oxygen-centered nucleophiles [5] to quinone methides, Sun and coworkers recently developed a series of conjugate additions employing other kind of nucleophiles. In 2016, they reported the first chiral phosphoric acid catalyzed enantioselective addition of thiols to in situ generated ortho-quinone methides (o-QMs). The reaction condition was mild and moderate to good efficiencies were obtained (Scheme 1) [6].
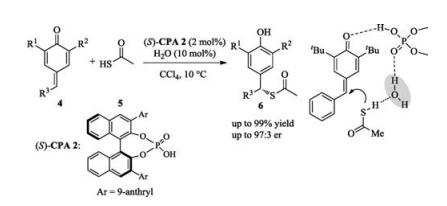
Soon after that, the same group published a chiral phosphoric acid catalyzed asymmetric 1, 6-addition of naphthols to in situ generated para-quinone methides (p-QMs) [7]. In 2016, Li reported an asymmetric 1, 6-conjugate addition of para-quinone methides using thioacetic acid 5 as nucleophile. Theoretical studies indicated that a water-bridged proton transfer is a potentially favorable reaction pathway (Scheme 2) [8].
In contrast with their oxygen-containing analogous ortho-quinone methides, aza-ortho-quinone methides are rarely employed in asymmetric reactions. In 2016, Rueping reported an asymmetric 1, 4-addition of thiols and alcohols to aza-ortho-quinone methides. In this work, the authors provided a reliable method for the synthesis of potent drug candidates which previously are not easily accessible (Scheme 3) [9].
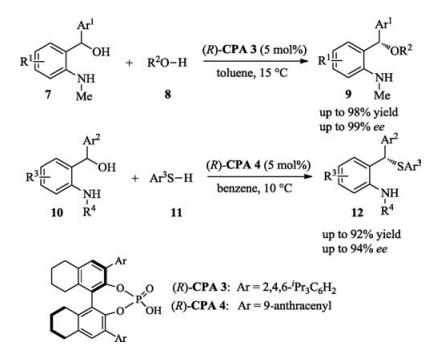
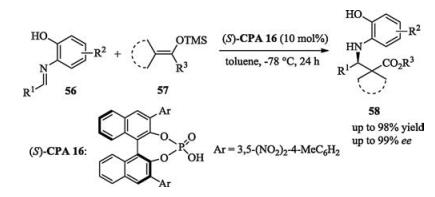
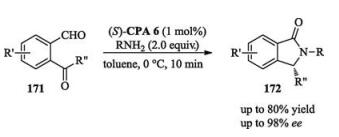
Due to the low nucleophilicity of N—H motif, the conjugate addition of the N—H in indoles and carbazoles to quinone methide analogues is more difficult than other hetero-atom-centered nucleophiles. After screening of a series of different protecting groups, Sun found that by employing a chiral phosphoric acid as catalyst, bulky 1-adamantanecarbonyl protected 4-amino-benzyl alcohols 13 reacted with 2, 3-disubstituted indoles or carbazoles smoothly to afford desired products with excellent enantioselectivities (up to 99% ee, Scheme 4) [10].

Hantzsch esters are regarded as hydride surrogate which reduce a series of organic compounds. Sun and coworkers recently realized the asymmetric reduction of 4-hydroyl benzyl tertiary alcohols to corresponding methine derivatives using chiral phosphoric acid as catalyst and Hantzsch ester as reductant. It should be noted that, after mechanism studies, they fully disclosed the actual role of the additive boronic acid through the whole reaction process (Scheme 5) [11].
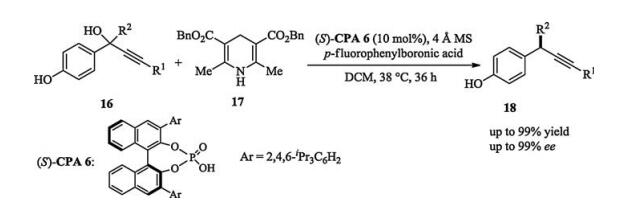
In addition to the widely reported 1, 4 and 1, 6-conjugate addition, Sun and coworkers recently demonstrated a synthesis of chiral tetrasubstituted allenes via a rarely reported 1, 8-conjugate addition of para-quinone methides. This process characterized by the construction of axial chirality from quinone methides (Scheme 6) [12].
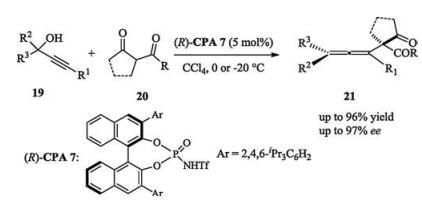
Studies on the conjugate addition of carbon-centered nucleophiles to quinone methides and aza-quinone methides were also reported by other groups [13].
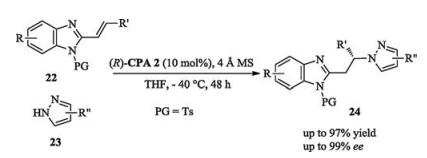
In 2016, Terada reported a highly enantioselective aza Michaeltype addition reactions between N-protected alkenyl benzimidazoles and either pyrazoles or indazoles. Structural analysis of the transition states revealed that both the catalyst substituent and the N-protective group of benzimidazole are essential to obtain high enantioselectivity (Scheme 7) [14].
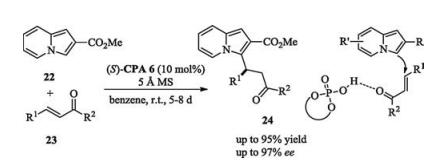
List and Coelho recently reported a catalytic asymmetric conjugate addition of indolizines to α, β-unsaturated ketones (Scheme 8) [15]. Mechanistically, the reaction proceeded via a monofunctional activation mode, because indolizines lack an available NH moiety to bond with the basic moiety of chiral phosphoric acid catalyst via hydrogen-bonding.
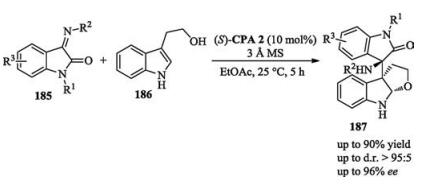
You recently reported a chiral phosphoric acid catalyzed intramolecular dearomative Michael addition of indoles to enones. This method provided spiro-indolenines bearing a quaternary stereogenic center with good yields and enantioselectivities (Scheme 9) [16].

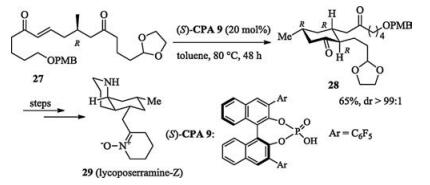
Chiral phosphoric acid catalyzed intramolecular Michael addition was successfully applied in the total synthesis of lycoposerramine-Z. The enantioselective construction of the multifuctionalized cyclohexanone was the initial step of the whole process (Scheme 10) [17].
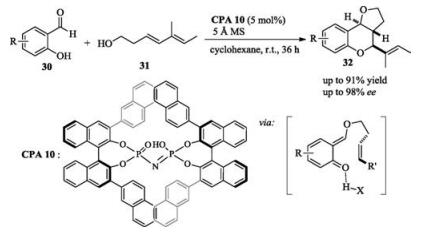
In addition to Michael addition, o-QMs have a high propensity to undergo [4 + 2] hetero-Diels-Alder reaction with dienophiles. In 2017, List reported an asymmetric intramolecular [4 + 2] cycloaddition of in situ generated ortho-quinone methides catalyzed by a confined chiral imidodiphosphoric acid catalyst. Through the reaction pathway, salicylaldehydes first react with dienyl alcohols to give transient ortho-quinone methide intermediates, which then undergo an intramolecular [4 + 2] cycloaddition to afford the highly functionalized final products with perfect diastereoselectivity and enantioselectivity (Scheme 11) [18].
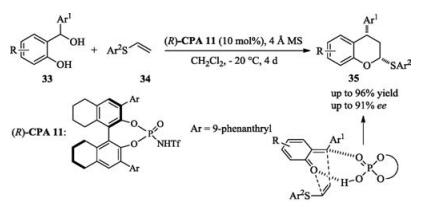
Sun recently reported a catalytic asymmetric [4 + 2] cycloaddition of ortho-quinone methides and vinyl sulfides (Scheme 12) [19]. Mechanistically, the reaction is a new example of the rarely achieved sole activation of o-QM for asymmetric control. Meanwhile, other groups also reported the works on asymmetric [4 + 2] cycloaddition of ortho-quinone methide derivatives [20].
Besides o-QMs, other kinds of dienes also undergo [4 + 2] cycloaddition. In 2016, Schneider reported an aza-ortho-quinone methides involved hetero-Diels-Alder reaction for the enantioselective synthesis of chiral tetrahydroacridines, which are privileged structural motifs exhibiting a broad range of bioactivities (Scheme 13) [21].
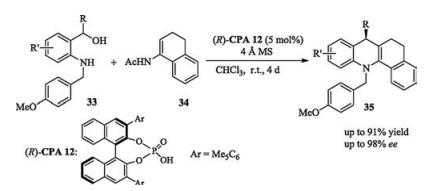
In 2016, Masson reported a catalytic asymmetric oxidative nitroso-Diels-Alder reaction of N-arylhydroxylamines. The dienophiles-arylnitroso derivatives were generated in situ by the oxidation of mCPBA (Scheme 14) [22]. Similar to this strategy, Masson reported an oxidative [3 + 2] cycloaddition using 1 equiv. of PIDA (phenyliododiacetate) as oxidant to generate the 3π partners (quinones from hydroquinones) in situ [23].
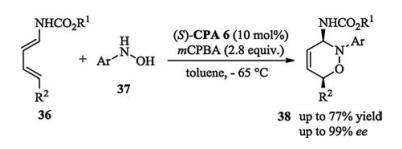
Intermolecular and intramolecular catalytic asymmetric Povarov-type reactions were developed by Shi and Masson independently in 2016 (Scheme 15) [24]. In Shi's three-component Povarov protocol, upon the activation of a chiral phosphoric acid, 3- methyl-2-vinylindoles reacted with the in situ generated aldimines to furnish tetrahydroquinoline motifs with excellent yields and stereoselectivities. In Masson's work, the catalyst loading could be lowered to 1 mol% and the desired product could be obtained in pure form without column chromatography.
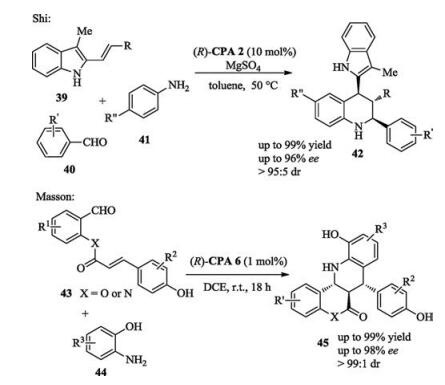
A new type of chiral phosphoric acid containing two different acidic sites was designed by Momiyama and Terada for a catalytic asymmetric [4 + 2] cycloaddition of Azopyridinecarboxylate and Amidodienes. Mechanistic study revealed that the two acidic sites in the catalyst cooperatively functioned to result in excellent catalytic activity and stereocontrol (Scheme 16) [25].

An enantioselective oxidative [4 + 3] cycloadditions between allenamides and furans was reported by Uria and Vicario. DMSO was used as an oxidant to generate the oxyallyl cations which coupled with furans (Scheme 17) [26]. Mechanistically, a bifunctional mode of activation combined hydrogen bonding with ion pair was proposed.

In 2017, Shi reported a chiral phosphoric acid catalyzed asymmetric [3 + 2] cycloaddition of 2-indolylmethanols with phydroxystyrenes (Scheme 18) [27]. Mechanically, a dual hydrogenbonding activation mode of the chiral catalyst toward the two substrates was proven to be crucial in this reaction.

Recently, catalytic stereoselective 1, 3-dipolar cycloaddition [28], [3 + 2] [29], [3 + 3] [30], [4 + 2] [31], and [4 + 3] [32] cycloadditions were also developed by Shi and others.
In 2016, Terada reported a chiral phosphoric acid catalyzed diastereo- and enantioselective Mannich-type reaction between enamides and thiazolones. Based on this method, a series of Mannich adducts were efficiently obtained and could be further transformed into ring opening products (Scheme 19) [33].

In 2016, Zhou and Yamamoto developed a new BINOL-derived chiral phosphoric acid to efficiently catalyze Mukaiyama–Mannich reaction. Perfect enantioselectivity (ee up to >99%) and diastereoselectivity (syn/anti up to >99/1) were achieved. The introduction of nitro group into the catalysts can enhance their acidity, benefiting from which, both the yields and enantioselectivities were improved in this case (Scheme 20) [34].
In 2016, Toste reported a chiral phosphoric acid catalyzed enantioselective synthesis of flavonoids. In this work, the authors designed a biomimetic chiral anion phase-transfer approach through which an insoluble benzopyrylium cation combined with a phosphoric acid anion to generate a soluble chiral benzopyrylium ion pair (Scheme 21) [35].

In 2016, Tan published an important work on the asymmetric synthesis of SPINOL derivatives, key intermediate compounds of a series of other chiral ligands and chiral phosphoric acids themselves. This chemistry gave a highly efficient approach to SPINOLs with good yield and excellent enantioselectivities under the catalysis of a SPINOL-derived chiral phosphoric acid. It was a practical method that the catalyst loading could be low to 0.1 mol% in scale-up synthesis. Interestingly, the absolute configuration of the product SPINOLs were opposite to that of the catalyst (Scheme 22) [36].
In 2016, Shao developed a catalytic asymmetric Friedel–Craftstype arylation reaction of C-alkynyl imines. From C-alkynyl N-Bocprotected N, O-acetals and electron-deficient phenols, a series of enantioenriched propargylamines were obtained with good yields and enantioselectivities (Scheme 23) [37].

In 2016, Lin reported a chiral phosphoric acid catalyzed enantioselective aza-Friedel-Crafts reaction of trifluoromethyl benzoxazinones with pyrroles (Scheme 24) [38]. Mechanistic studies and theory calculations suggested that triple-hydrogenbonding interactions were involved in the transition structure.

In 2017, Shi reported an efficient synthesis of axially chiral biaryl compounds via chiral phosphoric acid catalyzed asymmetric coupling reactions of 2-naphthols with 2-indolylmethanols. This chemistry exhibited good synthetic potential in the development of new types of chiral catalysts or ligands (Scheme 25) [39].Meanwhile, the same group published a supplementary work using different nucleophiles [40].

In 2017, Samanta and Yamamoto reported an asymmetric bromocyclization of polyenes using 1, 3-dibromo-5, 5-dimethylhydantoin as bromonium ion source and a chiral chiral phosphoric acid as catalyst. This is a practical way to natural product synthesis (Scheme 26) [41]. Besides, other groups also published beautiful works on the catalytic asymmetric Friedel–Crafts reactions [42].

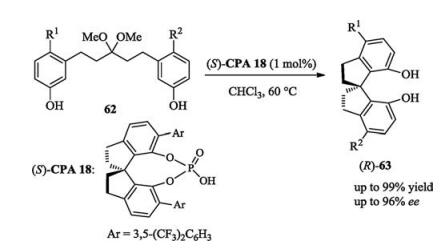
Compared with its counterpart oxirane ring, the low ringopening propensity of the four-membered oxetane ring makes it more challenging to react with nucleophilic reagents. After early successful attempt to open the oxetane ring under chiral phosphoric acid catalysts to generate chiral products [43], Sun group successively reported two works on the enantioselective oxetane ring opening reactions employing intramolecular and intermolecular nucleophiles respectively.
In their intramolecular nucleophilic attack case, upon suitable activation by an appropriate chiral phosphoric acid catalyst (R)- CPA 5, a series of oxetane derivatives underwent stereocontrolled oxetane ring opening reaction to form chiral 1, 4-dioxanes bearing quaternary stereocenters (Scheme 27) [44].

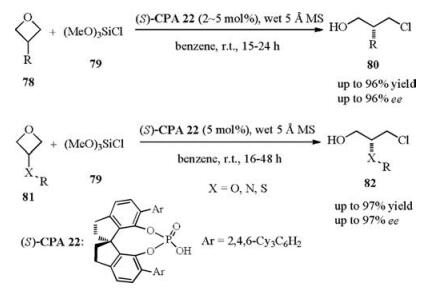
In their more challenge intermolecular nucleophilic attack case, a new catalyst chiral phosphoric acid (S)-CPA22 was developed for the desired transformation with high enantiomeric excesses. More importantly, the unprecedented use of wet molecular sieves for controlled HCl release was proved to be essential for the excellent enantiocontrol. Furthermore, the ring opening products can be further transformed to other products by simple substitution with different nucleophiles, which cannot open the oxetane ring directly (Scheme 28) [45].
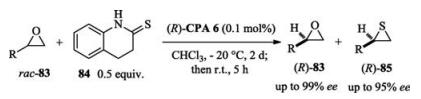
In 2016, List reported a kinetic resolution process to obtain enantio-enriched thiiranes and epoxides from racemic epoxides. Using thiourea as sulfur donor and (R)-CPA6 as catalyst, the reaction system was so efficient that the catalyst loadings can be low to 0.01 mol % (Scheme 29) [46].
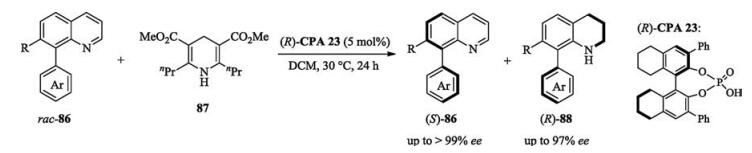
Later on, Zhou developed an asymmetric transfer hydrogenation reduction for the kinetic resolution of axially chiral substituted quinolines. Using chiral phosphoric acid as catalyst and Hantzsch ester as reductant, two kinds of axially chiral compounds were obtained with up to 209 of selectivity factor (Scheme 30) [47].
In the same year, Akiyama developed a strategy for the oxidative kinetic resolution of racemic indolines. Under a chiral phosphoric acid catalyst, the in situ formed iminium intermediate was hydrogenated by the same enantiomer of indoline to afford another enantiomer of indoline by a self-redox mechanism (Scheme 31) [48].

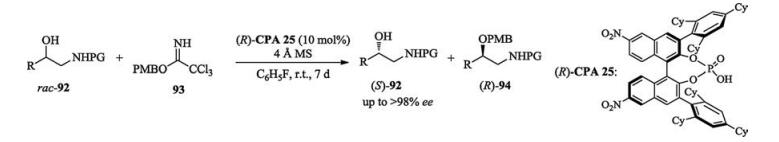
A kinetic resolution of β-amino alcohols by p-methoxybenzylation was reported by Yamada and Takasu. Using a chiral alkylating agent generated in situ from trichloroacetimidate and a chiral phosphoric acid, both enantioenriched alkylated chiral β-amino alcohols and one enantiomer of the substrate alcohols were obtained with high yields and enantioselectivities (Scheme 32) [49].
In 2017, List reported a kinetic resolution of primary amines via carbonyl-amine condensations. Under the catalysis of a chiral phosphoric acid, 1, 3-diketones reacted with racemic α-branched primary amines to give the enantioenriched enaminones and one enantiomers of the amine starting materials (Scheme 33) [50].

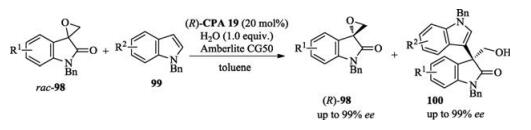
In 2017, Hong and Wang reported a kinetic resolution of spiroepoxyoxindoles based on an asymmetric Friedel–Crafts alkylation of indoles. This chemistry provided two kinds of useful compounds with good yields and enantioselectivities. Excellent s factors were obtained in all cases (Scheme 34) [51].
In 2017, Shimoda and Yamamoto reported a kinetic resolution of 2-pyridyl esters via amide bond formation catalyzed by a chiral phosphoric acid. The key success factor of this kinetic resolution was that, esters bearing pyridine moiety could interact with a proton from chiral Brønsted acid to form a chiral ion pair, thus made the kinetic resolution possible (Scheme 35) [52].

In 2016, Akiyama group developed a chiral phosphoric acid catalyzed dynamic kinetic resolution strategy for the enantiodivergent atroposelective synthesis of chiral biaryl compounds. By the proper choice of hydroxyaniline derivatives, biaryl lactols preceded a reductive amination reaction to furnish both R and S isomers of the chiral biaryl products with excellent yields and enantioselectivities (Scheme 36) [53].
A dynamic kinetic resolution was reported by Wang and Zhu in 2016. In the presence of a chiral phosphoric acid, isonitriles reacted with racemic isobenzofuranone derivatives smoothly to afford enantio-enriched isoindolinone products with good yields and enantioselectivities. In this work, the authors also developed an enantioselective three-component Ugi reaction to yield the same kind of products (Scheme 37) [54].
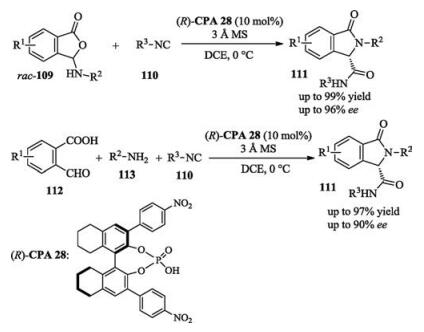
In the same year, Shi reported a catalytic asymmetric aminolysis of azlactone derivatives, which enabled the enantioselective dynamic kinetic resolution of azlactones (Scheme 38) [55].

In 2016, List and co-authors reported a catalytic asymmetric oxa-Pictet-Spengler reaction catalyzed by a nitrated confined imidodiphosphoric acid catalyst. Based on this reaction, a series of enantio-enriched isochromans were synthesized in excellent yields from simple starting material (Scheme 39) [56].

Soon after that, List published two works on the catalytic asymmetric Prins cyclization to synthesize enantio-enriched substituted tetrahydropyrans and tetrahydrofurans, respectively. The authors supposed that a confined chiral catalyst microenvironment would be necessary for a successful enantioselective Prins cyclization. However, they found that their previously developed confined imidodiphosphate catalysts were insufficiently acidic to promote the asymmetric Prins cyclizations of unactivated aldehydes. This problem was finally addressed by designing a new type of catalysts, CPA 30 and CPA 31, in which the oxo group was replaced with a stronger electronacceptor, the NTf group (Scheme 40) [57].
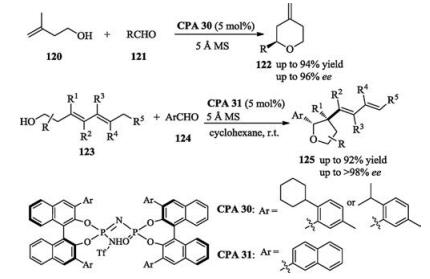
In 2016, Zhu reported an enantioselective vinylogous Pinacol rearrangement reaction. A chiral ion pair between the allylic cation and the chiral phosphoramide anion was postulated to result in the highly efficient chirality transfer (Scheme 41) [58]. Soon after that, an enantioselective acyloin rearrangement of α-hydroxy acetals was reported by the same group [59].

In the same year, List reported an enantioselective addition of allyltrimethylsilane to aldehydes catalyzed by a highly acidic imidodiphosphoric acid catalyst. This Hosomi–Sakurai reaction provided a series of chiral secondary homoallylic alcohols with excellent enantioselectivities (Scheme 42) [60].
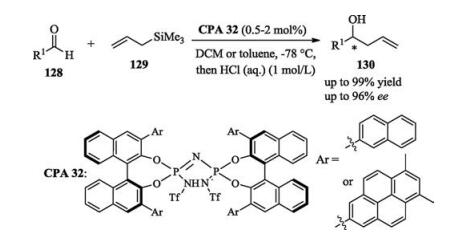
Two works on asymmetric Piancatelli rearrangement were reported by Rueping [61] and Sun [62] independently. The two groups both used anilines as nucleophiles but employed different chiral phosphoric acid catalysts (Scheme 43).

A chiral phosphoric acid catalyzed enantioselective reductive amination was developed by Rexit and Luo. This method used Hantzsch esters as reductants and provided a series of chiral 1- (pyridyl)-methyl amines as potentially useful chiral ligands (Scheme 44) [63].

An atroposelective synthesis of BINOL derivatives was reported by Sun, Kürti and Xu in 2016. In this work, a chiral phosphoric acid catalyzed cascade which involved sequential aminal-formation, sigmatropic rearrangement and rearomatization was developed to couple naphthols and iminoquinones in an enantioselective manner (Scheme 45) [64].

Later on, Tan reported a work on the atroposelective synthesis of axially chiral biaryl amino alcohol derivatives using 2- naphthylamine 141 as coupling partners (Scheme 46) [65].
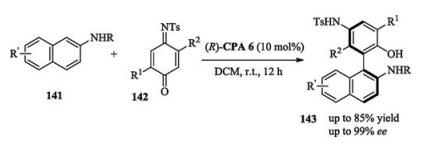
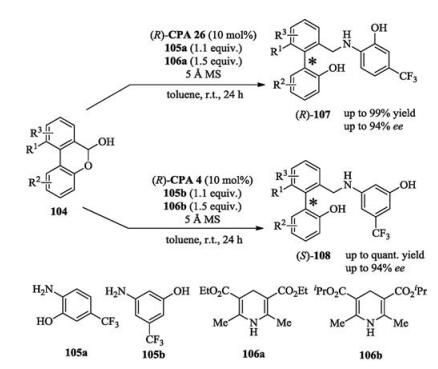
In 2016, Hong and Wang reported a chiral phosphoric acid catalyzed asymmetric oxidative dearomatizing coupling between naphthols and quinones. In this reaction, 3 equiv. of quinones were used as coupling partners as well as oxidants (Scheme 47) [66]. Later on, Chen and Zhou developed a non-oxidation strategy which used quinone monoimides as coupling partners (Scheme 47) [67].

A chiral phosphoric acid catalyzed asymmetric thioacetalization was reported by Zhou in 2016. This is the first example of catalytic asymmetric thioacetalization of salicylaldehyde and dithiol (Scheme 48) [68].

In 2016, Toste reported an asymmetric addition of α-branched cyclic ketones to allenamides. The adducts could be either hydrolyzed to give a formal Michael addition product (1, 5-keto aldehydes) or be transformed to 1, 4-keto aldehydes by the oxidative cleavage of enamide moiety. Both of the aldehydes are important building blocks in organic synthesis (Scheme 49) [69].

An asymmetric transfer hydrogenation of quinolone derivatives was reported by Jiang in 2016. In the presence of a chiral phosphoric acid and a Hantzsch ester, a series of chiral 2, 3- disubstituted 1, 2, 3, 4-tetrahydroquinoline derivatives were obtained in good yields and with excellent enantioselectivities (Scheme 50) [70].

In 2017, Zhu developed a method for the synthesis of chiral vicinal diamines via nucleophilic addition of hydrazones to imines. The chemo-, diastereo-, and enantioselectivity were all excellent and the adducts can be easily transformed into monoprotected or free diamines (Scheme 51) [71].
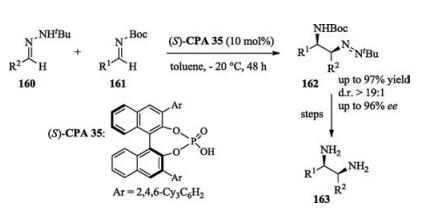
Earlier in the same year, Masson also reported an enantioselective three-component reaction to access chiral vicinal diamines [72].
2017, You reported a chiral phosphoric acid catalyzed asymmetric Pictet–Spengler reaction of indolyl dihydropyridines. The reaction condition was mild and the enantioenriched products tetrahydro-β-carbolines were obtained in good to excellent yields. The authors also exhibited the great synthetic potential of this method in the total synthesis of natural products (Scheme 52) [73].
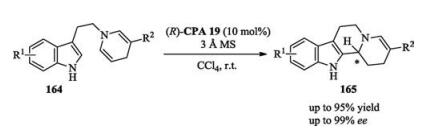
Later on, You reported an asymmetric fluorinative dearomatization of tryptamines catalyzed by chiral phosphoric acid. Proton sponge was added to neutralize the HBF4 generated in situ, because the latter would result in a racemic background reaction (Scheme 53) [74].
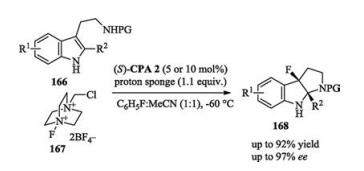
Recently, Zhang and Zu reported a chiral phosphoric acid catalyzed enantioselective aza-pinacol rearrangement. This reaction was applied to the asymmetric synthesis of a key intermediate of natural product (Scheme 54) [75].
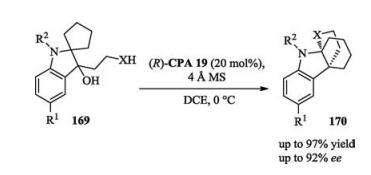
Very recently, Seidel reported a catalytic enantioselective synthesis of isoindolinones through the condensation of 2- acylbenzaldehydes and anilines. This chemistry featured low catalyst loading and very short reaction time (Scheme 55) [76].
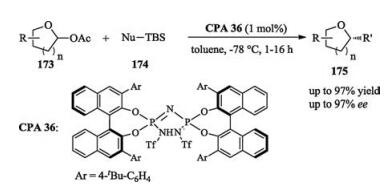
In 2107, List reported an asymmetric nucleophilic substitution between lactol acetates and enol silanes. Non-aromatic, cyclic oxocarbenium ions are difficult to interact with catalyst, making the asymmetric nucleophilic substitutions of them difficult to control. The authors employed a newly developed high-acidic, confined imidodiphosphorimidate catalyst CPA 36, whose highly stabilized counteranion formed an ion pair with the cyclic oxocarbenium ion to enable the highly enantioselective synthesis of substituted oxygen heterocycles (Scheme 56) [77].
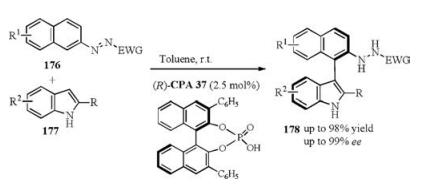
Tan recently reported a chiral phosphoric acid catalyzed asymmetric arylation of indoles. In this work, the azo group can effectively act as both a directing and activating group for organocatalytic asymmetric arylation of indoles, affording a wide range of axially chiral arylindoles with good yields and excellent enantioselectivities (Scheme 57) [78].
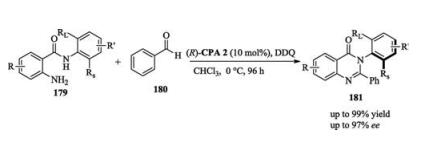
As part of their work to construct axially chiral compounds, Tan recently developed a chiral phosphoric acid catalyzed enantioselective approach to axially chiral arylquinazolinones. In this reaction, aldehyde 180 reacted with aniline 179 to generate an aminal intermediate, which then underwent dehydrogenation in the presence of an oxidant to furnish the final product (Scheme 58) [79].
In 2017, Huang reported an enantioselective synthesis of 1, 3- disubstituted 1, 3-dihydroisobenzofurans catalyzed by a chiral phosphoric acid. The whole process involved an asymmetric allylboration/oxo-Michael reaction cascade. Although the dr values were from 2.5:1 to 1.1:1, both the cis- and trans-isomers were obtained with high enantioselectivities (Scheme 59) [80].

In 2017, Shi reported a catalyst-controlled chemoselective and enantioselective reactions. As part of this work, a diastereoselective and enantioselective dearomative cyclization reaction was developed in the presence of a chiral phosphoric acid (Scheme 60) [81].
Chiral phosphoric acids are a series of catalysts with diverse substituents on the different backbones, thus generating a broad range of acidities and various kinds of chiral pockets. They have emerged as one of the fastest growing fields of organic catalysts and will continue to be a research hot-topic in the future. Firstly, we anticipate more and more new types of these catalysts will be designed. Based on those catalysts, new reaction modes will be developed to activate more substrate classes such as unactivated carbonyl compounds or simple olefins. Secondly, with the developments in chiral phosphoric acid catalyzed multicomponent and cascade reactions, chiral phosphoric acid catalyzed reactions will become reliable tools in the synthesis of more complex molecules such as natural products or pharmaceuticals. Finally, although chiral phosphoric acids are intensely studied in academia, it is anticipated that they will be widely employed in industry in the near future.
Financial support from the National Natural Science Foundation of China (No. 21772046), the Recruitment Program of Global Experts (1000 Talents Plan), the Natural Science Foundation of Fujian Province (No. 2016J01064), and Program of Innovative Research Team of Huaqiao University (No. Z14X0047) are gratefully acknowledged.
T. Akiyama, J. Itoh, K. Yokota, K. Fuchibe, Angew. Chem. Int. Ed. 43(2004) 1566-1568. doi: 10.1002/(ISSN)1521-3773
D. Uraguchi, M. Terada, J. Am. Chem. Soc. 126(2004) 5356-5357. doi: 10.1021/ja0491533
(a) T. Akiyama, Chem. Rev. 107 (2007) 5744-5758;
(b) A. Zamfir, S. Schenker, M. Freund, S. B. Tsogoeva, Org. Biomol. Chem. 8 (2010) 5262-5276;
(c) M. Terada, Synthesis (2010) 1929-1982;
(d) J. Yu, F. Shi, L. Z. Gong, Acc. Chem. Res. 44 (2011) 1156-1171;
(e) M. Rueping, A. Kuenkel, I. Atodiresei, Chem. Soc. Rev. 40 (2011) 4539-4549;
(f) M. Terada, Bull. Chem. Soc. Jpn. 83 (2010) 101-119.
(a) W. Zhao, Z. Wang, B. Chu, J. Sun, Angew. Chem. Int. Ed. 54 (2015) 1910-1913;
(b) Y. F. Z. Wang, J. Sun Wong, Angew. Chem. Int. Ed. 54 (2015) 13711-13714.
Z. Lai, Z. Wang, J. Sun, Org. Lett. 17(2015) 6058-6061. doi: 10.1021/acs.orglett.5b03072
Z. Lai, J. Sun, Synlett 27(2016) 555-558. doi: 10.1002/chin.201628106/pdf
Y.F. Wong, Z. Wang, J. Sun, Org. Biomol. Chem. 14(2016) 5751-5754. doi: 10.1039/C6OB00125D
N. Dong, Z.P. Zhang, X.S. Xue, X. Li, J.P. Cheng, Angew. Chem. Int. Ed. 55(2016) 1460-1464. doi: 10.1002/anie.201509110
A. Chatupheeraphat, H.H. Liao, S. Mader, et al., Angew. Chem. Int. Ed. 55(2016) 4803-4807. doi: 10.1002/anie.201511179
M. Chen, J. Sun, Angew. Chem. Int. Ed. 56(2017) 4583-4587. doi: 10.1002/anie.201701947
M. Chen, J. Sun, Angew. Chem. Int. Ed. 56(2017) 11966-11970. doi: 10.1002/anie.201706579
D. Qian, L. Wu, Z. Lin, J. Sun, Nat. Commun. 8(2017) 567. doi: 10.1038/s41467-017-00251-x
(a) G. Li, H. Liu, Y. Wang, et al., Chem. Commun. 52 (2016) 2304-2306;
(b) Q. Wu, C. Ma, X. H. Du, et al., Tetrahedron: Asymmetry 27 (2016) 307-316.
Y.Y. Wang, K. Kanomata, T. Korenaga, M. Terada, Angew. Chem. Int. Ed. 55(2016) 927-931. doi: 10.1002/anie.201508231
J.T.M. Correia, B. List, F. Coelho, Angew. Chem. Int. Ed. 56(2017) 7967-7970. doi: 10.1002/anie.201700513
Y. Zhou, Z.L. Xia, Q. Gu, S.L. You, Org. Lett. 19(2017) 762-765. doi: 10.1021/acs.orglett.6b03610
L.D. Zhang, L.R. Zhong, J. Xi, X.L. Yang, Z.J. Yao, J. Org. Chem. 81(2016) 1899-1904. doi: 10.1021/acs.joc.5b02723
Y. Xie, B. List, Angew. Chem. Int. Ed. 56(2017) 4936-4940. doi: 10.1002/anie.201612149
Z. Wang, J. Sun, Org. Lett. 19(2017) 2334-2337. doi: 10.1021/acs.orglett.7b00867
(a) T. Hodík, C. Schneider, Org. Biomol. Chem. 15 (2017) 3706-3716;
(b) C. Gharui, S. Singh, S. C. Pan, Org. Biomol. Chem. 15 (2017) 7272-7276;
(c) L. Zhang, Y. Liu, K. Liu, et al., Org. Biomol. Chem. 15 (2017) 8743-8747;
(d) T. Z. Li, C. A. Geng, X. J. Yin, et al., Org. Lett. 19 (2017) 429-431;
(e) K. Gebauer, F. Reuß, M. Spanka, C. Schneider, Org. Lett. 19 (2017) 4588-4591.
M. Kretzschmar, T. Hodik, C. Schneider, Angew. Chem. Int. Ed. 55(2016) 9788-9792. doi: 10.1002/anie.201604201
A. Dumoulin, G. Masson, J. Org. Chem. 81(2016) 10154-10159. doi: 10.1021/acs.joc.6b01256
C. Gelis, M. Bekkaye, C. Lebe'e, F. Blanchard, G. Masson, Org. Lett. 18(2016) 3422-3425. doi: 10.1021/acs.orglett.6b01593
(a) W. Dai, X. L. Jiang, J. Y. Tao, F. Shi, J. Org. Chem. 81 (2016) 185-192;
(b) L. Jarrige, F. Blanchard, G. Masson, Angew. Chem. Int. Ed. 56 (2017) 10573-10577.
N. Momiyama, H. Tabuse, H. Noda, et al., J. Am. Chem. Soc. 138(2016) 11353-11359. doi: 10.1021/jacs.6b07150
L. Villar, U. Uria, J.I. Martinez, et al., Angew. Chem. Int. Ed. 56(2017) 10535-10538.
M.M. Xu, H.Q. Wang, Y. Wan, S.L. Wang, F. Shi, J. Org. Chem. 82(2017) 10226-10233. doi: 10.1021/acs.joc.7b01731
(a) Y. M. Wang, H. H. Zhang, C. Li, T. Fan, F. Shi, Chem. Commun. 52 (2016) 1804-1807;
(b) Z. Zhang, W. Sun, G. Zhu, et al., Chem. Commun. 52 (2016) 1377-1380.
(a) Y. Wang, Q. Wang, J. Zhu, Chem. -Eur. J. 22 (2016) 8084-8088;
(b) X. X. Sun, H. H. Zhang, G. H. Li, L. Meng, F. Shi, Chem. Commun. 52 (2016) 2968-2971;
(c) M. Zhang, S. Yu, F. Hu, et al., Chem. Commun. 52 (2016) 8757-8760;
(d) T. Fan, H. H. Zhang, C. Li, Y. Shen, F. Shi, Adv. Synth. Catal. 358 (2016) 2017-2031;
(e) Z. Q. Zhu, Y. Shen, X. X. Sun, et al., Adv. Synth. Catal. 358 (2016) 3797-3808.
(a) X. X. Sun, H. H. Zhang, G. H. Li, Y. Y. He, F. Shi, Chem. -Eur. J. 22 (2016) 17526-17532;
(b) X. X. Sun, C. Li, Y. Y. He, et al., Adv. Synth. Catal. 359 (2017) 2660-2670;
(c) C. Li, H. Lu, X. X. Sun, G. J. Mei, F. Shi, Org. Biomol. Chem. 15 (2017) 4794-4797.
C. Weilbeer, M. Sickert, S. Naumov, C. Schneider, Chem.-Eur. J. 23(2017) 513-518. doi: 10.1002/chem.201604356
G.J. Mei, Z.Q. Zhu, J.J. Zhao, et al., Chem. Commun. 53(2017) 2768-2771. doi: 10.1039/C6CC09775H
J. Kikuchi, N. Momiyama, M. Terada, Org. Lett. 18(2016) 2521-2523. doi: 10.1021/acs.orglett.6b00857
(a) F. Zhou, H. Yamamoto, Angew. Chem. Int. Ed. 55 (2016) 8970-8974;
(b) F. Zhou, H. Yamamoto, Org. Lett. 18 (2016) 4974-4977.
Z. Yang, Y. He, F.D. Toste, J. Am. Chem. Soc. 138(2016) 9775-9778. doi: 10.1021/jacs.6b05939
S. Li, J.W. Zhang, X.L. Li, D.J. Cheng, B. Tan, J. Am. Chem. Soc. 138(2016) 16561-16566. doi: 10.1021/jacs.6b11435
Y. Wang, L. Jiang, L. Li, et al., Angew. Chem. Int. Ed. 55(2016) 15142-15146. doi: 10.1002/anie.201608918
H. Lou, Y. Wang, E. Jin, X. Lin, J. Org. Chem. 81(2016) 2019-2026. doi: 10.1021/acs.joc.5b02848
H.H. Zhang, C.S. Wang, C. Li, et al., Angew. Chem. Int. Ed. 56(2017) 116-121. doi: 10.1002/anie.201608150
Y.Y. He, X.X. Sun, G.H. Li, G.J. Mei, F. Shi, J. Org. Chem. 82(2017) 2462-2471. doi: 10.1021/acs.joc.6b02850
R.C. Samanta, H. Yamamoto, J. Am. Chem. Soc. 139(2017) 1460-1463. doi: 10.1021/jacs.6b13193
(a) L. Wang, A. Rahman, X. Lin, Org. Biomol. Chem. 15 (2017) 6033-6041;
(b) S. G. Lee, S. G. Kim, RSC Adv. 7 (2017) 34283-34286;
(c) Y. Zhao, L. Wang, J. Zhao, Tetrahedron Lett. 58 (2017) 213-217.
(a) Z. Chen, B. Wang, Z. Wang, G. Zhu, J. Sun, Angew. Chem. Int. Ed. 52 (2013) 2027-2031;
(b) Z. Wang, Z. Chen, J. Sun, Angew. Chem. Int. Ed. 52 (2013) 6685-6688;
(c) Z. Chen, Z. Wang, J. Sun, Chem. -Eur. J. 19 (2013) 8426-8430.
W. Yang, J. Sun, Angew. Chem. Int. Ed. 55(2016) 1868-1871. doi: 10.1002/anie.201509888
W. Yang, Z. Wang, J. Sun, Angew. Chem. Int. Ed. 55(2016) 6954-6958. doi: 10.1002/anie.201601844
S. Liao, M. Leutzsch, M.R. Monaco, B. List, J. Am. Chem. Soc. 138(2016) 5230-5233. doi: 10.1021/jacs.6b01960
J. Wang, M.W. Chen, Y. Ji, S.B. Hu, Y.G. Zhou, J. Am. Chem. Soc.138(2016) 10413-10416. doi: 10.1021/jacs.6b06009
K. Saito, T. Akiyama, Angew. Chem. Int. Ed. 55(2016) 3148-3152. doi: 10.1002/anie.201510692
Y. Kuroda, S. Harada, A. Oonishi, et al., Angew. Chem. Int. Ed. 55(2016) 13137-13141. doi: 10.1002/anie.201607208
S. Das, N. Majumdar, C.K. De, et al., J. Am. Chem. Soc. 139(2017) 1357-1359. doi: 10.1021/jacs.6b12176
G. Zhu, G. Bao, Y. Li, et al., Angew. Chem. Int. Ed. 56(2017) 5332-5335. doi: 10.1002/anie.201700494
Y. Shimoda, H. Yamamoto, J. Am. Chem. Soc. 139(2017) 6855-6858. doi: 10.1021/jacs.7b03592
K. Mori, T. Itakura, T. Akiyama, Angew. Chem. Int. Ed. 55(2016) 11642-11646. doi: 10.1002/anie.201606063
Y. Zhang, Y.F. Ao, Z.T. Huang, et al., Angew. Chem. Int. Ed. 55(2016) 5282-5285. doi: 10.1002/anie.201600751
Y.C. Zhang, Q. Yang, X. Yang, Q.N. Zhu, F. Shi, Asian J. Org. Chem. 5(2016) 914-919. doi: 10.1002/ajoc.v5.7
S. Das, L. Liu, Y. Zheng, et al., J. Am. Chem. Soc. 138(2016) 9429-9432. doi: 10.1021/jacs.6b06626
(a) L. Liu, P. S. J. Kaib, A. Tap, B. List, J. Am. Chem. Soc. 138 (2016) 10822-10825;
(b) Y. Xie, G. J. Cheng, S. Lee, et al., J. Am. Chem. Soc. 138 (2016) 14538-14541.
H. Wu, Q. Wang, J. Zhu, Angew. Chem. Int. Ed. 55(2016) 15411-15414. doi: 10.1002/anie.v55.49
H. Wu, Q. Wang, J. Zhu, Angew. Chem. Int. Ed. 56(2017) 5858-5861. doi: 10.1002/anie.201701098
P.S.J. Kaib, L. Schreyer, S. Lee, R. Properzi, B. List, Angew. Chem. Int. Ed. 55(2016) 13200-13203. doi: 10.1002/anie.201607828
Y. Cai, Y. Tang, I. Atodiresei, M. Rueping, Angew. Chem. Int. Ed. 55(2016) 14126-14130. doi: 10.1002/anie.v55.45
H. Li, R. Tong, J. Sun, Angew. Chem. Int. Ed. 55(2016) 15125-15128. doi: 10.1002/anie.201607714
A.A. Rexit, S. Luo, M. Mailikezati, J. Org. Chem. 81(2016) 11384-11388. doi: 10.1021/acs.joc.6b01772
J.Z. Wang, J. Zhou, C. Xu, et al., J. Am. Chem. Soc. 138(2016) 5202-5205. doi: 10.1021/jacs.6b01458
Y.H. Chen, L.W. Qi, F. Fang, B. Tan, Angew. Chem. Int. Ed. 56(2017) 16308-16312. doi: 10.1002/anie.v56.51
G. Zhu, G. Bao, Y. Li, et al., Org. Lett 18(2016) 5288-5291. doi: 10.1021/acs.orglett.6b02609
X.Q. Li, H. Yang, J.J. Wang, et al., Chem.-Eur. J. 23(2017) 5381-5385. doi: 10.1002/chem.201701015
J.S. Yu, W.B. Wu, F. Zhou, Org. Biomol. Chem. 14(2016) 2205-2209. doi: 10.1039/C5OB02495A
X. Yang, F.D. Toste, Chem. Sci. 7(2016) 2653-2656. doi: 10.1039/C5SC04202J
J. Zhou, Q.F. Zhang, W.H. Zhao, G.F. Jiang, Org. Biomol. Chem. 14(2016) 6937-6941. doi: 10.1039/C6OB01176D
Y. Wang, Q. Wang, J. Zhu, Angew. Chem. Int. Ed. 56(2017) 5612-5615. doi: 10.1002/anie.201702295
A. Dumoulin, G. Bernadat, G. Masson, J. Org. Chem. 82(2017) 1775-1789. doi: 10.1021/acs.joc.6b03093
S.G. Wang, Z.L. Xia, R.Q. Xu, et al., Angew. Chem. Int. Ed. 56(2017) 7440-7443. doi: 10.1002/anie.201703178
X.W. Liang, C. Liu, W. Zhang, S.L. You, Chem. Commun. 53(2017) 5531-5534. doi: 10.1039/C7CC02419C
Y. Yu, J. Li, L. Jiang, J.R. Zhang, L. Zu, Angew. Chem. Int. Ed. 56(2017) 9217-9221. doi: 10.1002/anie.201705539
C. Min, Y. Lin, D. Seidel, Angew. Chem. Int. Ed. 56(2017) 15353-15357. doi: 10.1002/anie.201709182
S. Lee, P.S.J. Kaib, B. List, J. Am. Chem. Soc. 139(2017) 2156-2159. doi: 10.1021/jacs.6b11993
L.W. Qi, J.H. Mao, J. Zhang, B. Tan, Nat. Chem. 10(2017) 58-64. doi: 10.1038/nchem.2866
Y.B. Wang, S.C. Zheng, Y.M. Hu, B. Tan, Nat. Commun. 8(2017) 15489. doi: 10.1038/ncomms15489
X. Yang, S. Pang, F. Cheng, et al., J. Org. Chem. 82(2017) 10388-10397. doi: 10.1021/acs.joc.7b01856
F. Jiang, D. Zhao, X. Yang, et al., ACS Catal. 7(2017) 6984-6989. doi: 10.1021/acscatal.7b02279

Scheme 2 Enantioselective addition of thioacetic acids to para-quinone methides (p-QMs).

Scheme 3 Enantioselective addition of thiols and alcohols to aza-ortho-quinone methides.

Scheme 4 Enantioselective addition of indole and carbazoles to aza-para-quinone methides.

Scheme 8 Asymmetric Michael addition reactions of indolizines to α, β-unsaturated ketones.

Scheme 12 Asymmetric [4 + 2] cycloaddition of ortho-quinone methides and vinyl sulfides.

Scheme 24 Asymmetric aza-Friedel-Crafts reaction of trifluoromethyl benzoxazinones andpyrroles.

Scheme 36 Chiral phosphoric acid catalyzed dynamic kinetic resolution of biaryl lactols.

Scheme 37 Chiral phosphoric acid catalyzed dynamic kinetic resolution of isobenzofuranone.

Scheme 46 Atroposelective synthesis of axially chiral biaryl amino alcohol derivatives.

Scheme 47 Chiral phosphoric acid catalyzed asymmetric oxidative dearomatizing coupling.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
















































