Citation:
Mingqin Chang, Qian Xi, Ming Ji, Wenyuan Huang, Tingyu Gao, Yang Li. Synthesis and In Vitro Antitumor Activity of Novel Quinoxaline-Based Isoindolin-1-Ones[J]. Chemistry,
2021, 84(2): 154-161, 171.
Received Date:
03 July 2020 Accepted Date:
17 September 2020 Available Online:
18 February 2021
Abstract:
In the present investigation, a simple and facile synthesis of a series of structurally novel and intriguing quinoxaline-based isoindolin-1-ones through the one-step reaction of ethyl 3-bromomethylquinoxaline-2-carboxylatewith with arylamines or aliphatic amines in refluxing ethanol medium has been described. The preliminary screening for their in vitro anti-tumor activity against A549 and HT29 using the MTT assay revealed that halo-substituted (F, Cl, Br, I) compounds 7h~7k showed promising inhibitory activity, among which fluro-substituted 7h possessed the best activity against the two cell lines with the IC50 values of 1.50 and 3.77 μg/mL, respectively, which were higher than that of the reference drug cisplatin.
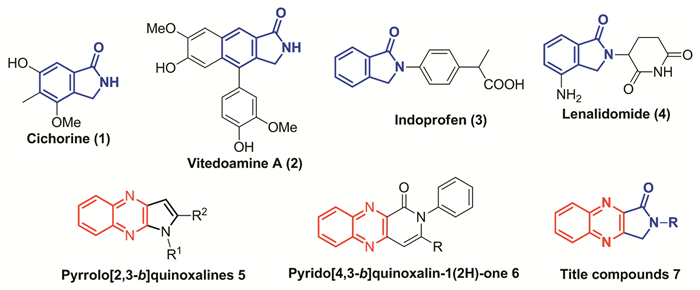
Isoindolin-1-one (or phthalimidine) is a core heterocyclic ring present in some naturally alkaloids like cichorine (1, Scheme 1)[1], vitedoamine A (2)[2], and also in some pharmacologically important synthetic compounds such as indoprofen (3)[3] and lenalidomide (4)[4]. The derivatives of this family have exhibited a broad spectrum of biological activi-ties, such as antimicrobial[5], anticancer[6], antinoci-ceptive[7]and antipsychotic properties[8]. Therefore, considerable synthetic efforts have been invested surrounding the isoindolin-1-one molecular template for further modification and functionalization to enhance the potency of this class of ring systems[9~11].
Scheme 1
Scheme 1.
生物活性的异吲哚啉-1-酮1~4、稠合喹喔啉5、6和目标化合物7的结构
Scheme 1.
Structures of bioactive isoindolin-1-ones 1~4, fused quinoxalines 5, 6 and the targeted compound 7
On the other hand, bioactive heterocycles when linked with quinoxaline moiety in fused or bonded forms usually results in novel hybrids with potent pharmacological properties[12~15]. Especially, the syntheses and anti-tumor activity of quinoxaline-based compounds have been frequently reported by both organic and medicinal chemists with the aim of developing new and effective anticancer agents[16~18]. For example, Keivanloo et al.[19] reported the Pd-Cu catalyzed synthesis of pyrrolo[2, 3-b]quinoxalines (5, Scheme 1). Hajri et al.[20]presented the synthesis and antitumor activity of tricyclic-fused pyrido[4, 3-b]quinoxalin-1(2H)-ones against A549 cell lines (6). Just recently, an advance on the synthesis and antitumoral activity of quinoxaline derivatives has been reviewed by Kaushal et al.[21]and Montana et al, respectively[22].
Keeping the above findings in mind and in view of molecular hybridization playing a prominent role in medicinal and combinatorial chemistry[23], it would be an attractive idea to replace the benzene moiety of isoindolin-1-one with quinoxaline ring to give rise to a new dimension of structural diversity as potential candidates for biological evaluation. Accordingly, in the context of our ongoing studies concerning the synthesis of new type of bioactive heterocyclic hybrids[24~27], we would like to report herein a simple and efficient synthesis of structurally novel and intriguing N-substituted-2, 3-dihydro-1H-pyrrolo [3, 4-b] quinoxalin-1-ones (7) as shown in Scheme 1. To the best of our knowledge, the synthesis of such meaningful compounds has not been achieved so far.
1.
Experimental
1.1
General
The chemical reagents used in this work were obtained from Energy Chemical and were used without purification. Ethyl 3-bromomethyl quinoxaline-2-carboxylate (8) was obtained by our recently reported method[26]. Melting points were determined by a WRS-1B melting point apparatus without correction. The 1H and 13C NMR spectra were recorded on an Agilent 400-NMR spectrometer using CF3COOH or CDCl3 as solvent. The reported chemical shifts (δ values) are given in parts per million downfield from tetramethylsilane (TMS) as the internal standard. HRMS (ESI) data were acquired on a Bruker Customer micrOTOF-Q 125 high resolution mass spectrometer with ESI. Elemental analyses were carried out on an EA 2400Ⅱ elemental analyzer (PerkinElmer, Waltham, MA). The progress of reactions was monitored by TLC on silica gel GF254 using ethyl acetate/petroleum ether (1∶4) as the eluent.
1.2
General procedure for the synthesis of 2-aryl/alkyl-2, 3-dihydro-1H-pyrrolo[3, 4-b]quinoxalin-1-ones (7a~7n)
A mixture of ethyl 3-(bromomethyl) quinoxaline-2-carboxylate (0.295 g, 1.0 mmol) and the respective amine (1.1 mmol) was refluxed in ethanol (20 mL). After the reaction (monitored by TLC), the mixture was cooled to room temperature and the solvent was removed under reduced pressure. The resulting precipitate was filtered off and washed with hexane to give the pure products 7a~7n. The yields, physical properties and the spectral and analytical data are given below.
1.3
Experimental procedure for in vitro antiproliferative activity test of target compounds on A549 and HT29 cell lines (MTT assay)
The A549 and HT29 cancer cell lines were cultured on cell culture flask using 2 mmol/L L-glutamine adjusted to contain 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mmol/L 4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) and 1.0 mmol/L sodium pyruvate in the Roswell Park Memorial Institute (RPMI) 1640 nutrient medium supplemented with 0.5 mg/mL G418 and 10% heat-inactivated fetal calf serum (FCS) (pH 7.2). The two cell types were seeded in 96-well culture plate at the cell density of 5×104 cells per well in 100 μL of culture medium at 37 ℃ in 5% CO2 incubator for 24 h seeding. The stock solutions of the test compounds 7 were prepared in DMSO. After incubation, the cells were treated with different concentrations of the tested compounds, made by serial dilution in culture medium, and incubated for 72 h with each concentration located three wells. Then the drug containing medium was removed and replaced by 100 μL fresh medium with 0.5 mg/mL MTT solution. After 4 h incubation, the medium with MTT was removed and 100 μL DMSO was added to each well. The plates were gently agitated until the color reaction was uniform. The OD values were measured using SPECTRA max 190Cell microplate reader under 490 nm (for absorbance of MTT formazan) and 630 nm (for the reference wavelength). All compounds were tested three times in each of the cell lines, and the results expressed as IC50 (inhibitory concentration 50%) were the averages of three determinations and calculated by using GraphPad Prism version 6.00 software from the non-linear curve.
2.
Results and Discussion
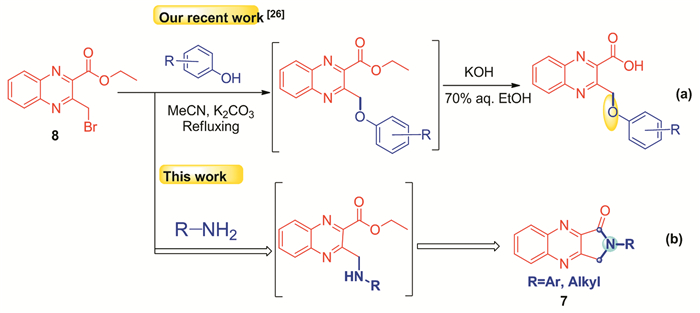
In recent years, a number of synthetic methods have been developed for the synthesis of isoindolin-1-one derivatives, including one-pot reaction of 2-formylbenzoic acid with amines and dimethyl phosphate followed by dephosphonylation reaction[28], palladium(0)-catalyzed carbonylation-amination[29], carbamoyl radical cyclization[30], Parham-type cyclization of iodinated benzyldicarbamates[31], condensation of anilines with o-phthalaldehydes[32], and inverse electron demand Diels-Alder cycloadditions[33]. Although these synthetic methodologies are effective to obtain isoindolin-1-ones, they are not easily extended to the synthesis of quinoxaline-based isoindolin-l-ones due to the unavailability of the required starting materials. Just recently, we have reported the preparation and application of ethyl 3-bromomethylquinoxaline-2-carboxylate (8) in the one-pot reaction with phenols for the flexible synthesis of a series of 3-aroxymethylquinoxaline-2-carboxylic acids as shown in Scheme 2[27]. Building on the evolving expertise, we envisioned that it might be possible to convert 8 into the desired quinoxaline-based isoindolin-l-one derivatives through a cascade nucleophilic substitution reaction with various amines followed by subsequent intramolecular C-N bond cyclization of the resulting intermediates in a single synthetic operation as outlined in Scheme 2.
Accordingly, the reaction of 8 with 1.1 equiv of aniline (9a) in refluxing MeCN was first investigated. As expected, the desired product 2-phenyl-2, 3-dihydro-1H-pyrrolo[3, 4-b]quinoxalin-1-one (7a) could be obtained but only in a low yield of 33% even after refluxing 24 h (Entry 1, Tab. 1). When using CH2Cl2 or CHCl3 as solvent, the reaction was fraught with difficulties associated with combination of starting materials and numerous products, from which the desired 7a could not separated in any appreciable yield (Entries 2 and 3, Tab. 1). Switching the solvent to acetone or tetrahydrofuran (THF) was found to be not suitable for the transformation as well, giving poor yields of highly impure products (Entries 4 and 5, Tab. 1). The attempt to use MeOH as solvent was also less satisfactory, and the expected product 7a was obtained in a moderate yield of 52% (Entry 6, Tab. 1). After these attempts, we were delighted to find that using EtOH as solvent could prompt the reaction proceeding efficiently with only one new compound being observed on TLC. When the reaction was completed within 10 h as monitored by TLC, the expected product 7a was obtained in a good yield of 76% (Entry 7, Tab. 1).
Scheme 2
Scheme 2.
基于喹喔啉结构的异吲哚啉-1-酮7的合成路线
Scheme 2.
The designed synthetic route for quinoxaline-based isoindolin-1-ones 7
Subsequently, the scope and limitations of the reaction were investigated by extending to various aromatic amines for building differently substituted analogs (as listed in Tab. 2). The results showed that the site of substituent present in the aromatic amines had an obvious steric hindrance effect on the product yields. For example, the reaction with o-toluidine scarcely proceeded, and the corresponding product 7b was detected only in negligible amount that did not warrant isolation (Entry 2, Tab. 2). The reaction with m-toluidine gave rise to 7c in a low yield of 41% (Entry 3, Tab. 2) even after refluxing 24 h. Only the p-toluidine reacted smoothly with 8, furnishing the product 7d in a good yield of 77% without any experimental difficulties (Entry 4, Tab. 2). Similarly, the reaction with 2-(tert-butyl)aniline scarcely proceeded and the starting materials were recovered unchanged (Entry 6, Tab. 2), while 4-(tert-butyl)aniline had a good reactivity with 8, affording 7g in a good yield of 76% (Entry 7, Tab. 2). Generally, various p-substituted aromatic amines with both electron-donating and slightly electron-withdrawing groups worked well, successfully giving the corresponding products in good yields.
Table 2
表 2
化合物7a~7k的合成及其体外抗A549和HT29肿瘤细胞活性
Table 2.
Synthesis and in vitro anti-tumor activity of the newly-synthesized compounds toward A549 and HT29
However, when using aromtic amines bearing strong electron-withdrawing groups such as NO2 and CN, the reaction scarcely proceeded and the starting materials were recovered unchanged. The possible reason might be that the strong electron-withdrawing groups render the amine highly electron-deficient and thus retard the reaction process. Due to the simplicity of the one-step reaction procedure and in order to diversify our work, we further extended the scope to some aliphatic amines. To our delight, these species such as methanamine, ethanamine and benzylamine were viable substrates for this transformation under the same reaction conditions, giving, not unexpectedly, the expected products 7l~7n in comparable yields of 67%, 70%, and 73%, respectively (Entries 12~14, Tab. 2). However, contrary to our expectation, the reaction with some secondary amines such as piperidine and piperazine did not proceed satisfactorily, giving highly impure mixtures of starting materials and numerous products. Work is currently ongoing and more studies toward extending the reaction scope will be part of our future efforts.
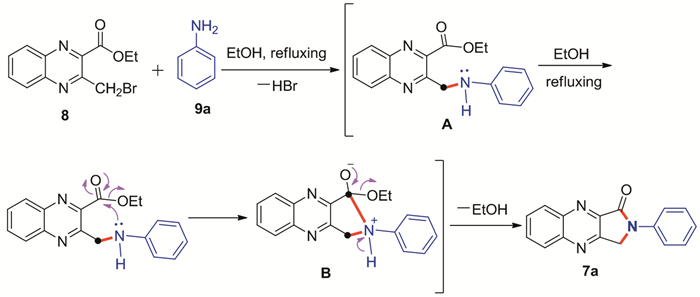
On the basis of these experiments, a mechanistic proposal portraying the probable sequence for the formation of the title compounds is outlined in Scheme 3, taking the formation of 7a as an example. Firstly, the nucleophilic substitution reaction of 8 with 9a produced the intermediate A, which undergoes subsequent intramolecular nucleophilic cyclization, resulting in a five-membered cyclic system B. Next, the elimination of EtOH in B leads to the formation of 7a. It is worthy to mention that an example that is particularly relevant to the present discussion was described in literature[34], wherein 7-isopropyl-1-methylazulen-4-amine underwent such intramolecular ring annulation reaction with methyl ester at 3-position to yield tricyclic-fused δ-lactams through the elimination of methanol without any catalyst.
Scheme 3
Scheme 3.
形成化合物7a可能的反应机理
Scheme 3.
Possible mechanistic pathway for the formation of product 7a
With these newly-synthesized quinoxaline-based isoindolin-1-ones in hand, we became interested in evaluating their anti-tumor activity. Accordingly, a primary screening for in vitro anti-tumor activity against human cancer cell lines A549 and HT29 were conducted by using a formazan dye (MTT) conversion assay. The results, as recorded in Tab. 2, revealed that the introduction of alkyl substituents such as methyl, ethyl, tert-butyl and benzyl groups as in products 7c~7e, 7g and 7l~7n displayed poor inhibitory activities (Entries 3~5, 7 and 12~14, Tab. 2). It shows that the alkyl group is not conducive to the inhibitory activity of the compounds. Interestingly, the introduction of halo group (F, Cl, Br and I) gave an important improvement of the inhibitory activity (Entries 8~11, Tab. 2), increasing the anti-tumor potential with the order of F>Br>I>Cl. Especially, the fluoro-substituted 7h (Entry 8, Tab. 2) had the best activity with the IC50 values of 1.50 and 3.77 μg/mL against A549 and HT29, being better than the reference drug Cisplatin. These insights from the in vitro anti-tumor activity might provide valuable information for further optimization of the series of derivatives, and hopefully, contribute to the development of new and effective anti-tumor candidates. Our next efforts will mainly focus on the structural activity relationship study through structural optimization and exploring more structural diversity towards the ultimate goal of providing intriguing lead compounds for current anti-tumor activity studies.
3.
Conclusion
In summary, we have achieved a concise and facile synthesis of a series of new quinoxaline-based isoindolin-1-ones. The synthetic protocol described here might be attractive as it is simple and does not involve the use of expensive reagents or catalysts. The preliminary in vitro anti-tumor activity bioassay showed that compounds with halogen groups (7h~7k) had higher inhibitory activity, and fluoro-substituted 7h showed the best activity with the IC50 values up to 1.50 μg/mL against A549 and 3.77 against HT29, being comparable with the reference Cisplatin. These results might give an important insight to future optimization of the series of quinoxaline derivatives. Work is currently ongoing, mainly focusing on the anti-tumor mechanism study, structural optimization and further application of this method for structurally diverse derivatives, which will be communicated in due course.
[1]
Stierle A, Hershenhorn J, Strobel G. Phytochemistry, 1993, 32(5): 1145~1149.
[2]
Ono M, Nishida Y, Masuoko C, et al. J. Nat. Prod., 2004, 67(12): 2073~2075.

 下载:
下载:



 下载:
下载:




