图式 1.
目标化合物的分子结构
Scheme 1.
The structures of target molecules

具有扭曲的分子内电荷转移(TICT)结构特性的分子在激发态时能围绕单键旋转并发生能量转移或电荷转移,具有Stokes位移大、荧光发射光谱对环境敏感的特点,在有机发光二极管(OLED)、光学传感器、光伏器件等领域显示出潜在的应用前景[1]。通过调节中心共轭基团的结构,在化合物中引入TICT特性,能显著改变其电荷离域特征与电荷转移性质,影响分子的光物理性质与聚集状态[2]。分子内电荷转移与共轭特性对线性吸收与双光子吸收有着显著影响[3]。其中双光子吸收(2PA)是指在激光照射下物质的一个分子同时吸收两个光子到达激发态的非线性光学过程[4],由于其在荧光成像领域具有分辨率高以及空间穿透性好等优点,广泛应用于生物荧光成像[5]、3D微结构制造[6]、光动力治疗等领域[7]。由于三苯胺基团内较强的耦合作用,同时芳香酮类小分子具有合成简便、易于修饰的优点,本文设计合成了一系列以二苯胺为电子给体、芳香酮基团为电子受体的D-π-A-π-D型共轭化合物(见图式 1),通过表征线性与双光子吸收性能,研究了分子的共轭结构以及取代基位置对其电荷转移性质的影响,并讨论了化合物在双光子荧光成像方面的应用潜力。
所有合成原料均购自百灵威和伊诺凯公司,所有试剂均采用常规方法纯化。紫外可见吸收光谱通过Hitachi U3900光谱仪测试;荧光发射光谱通过Hitachi F4500光谱仪测试。荧光量子产率使用若丹明B的甲醇溶液作为参比[8]。1H NMR和13C NMR谱通过Bruker Avance-DPX400核磁共振谱仪测试。高分辨质谱(HRMS)通过Waters GCT Premier质谱仪测试。所有的测试均在室温条件下完成。
所有化合物均通过Suzuki偶联反应偶联相应的二溴取代芳基酮和4-硼酸三苯胺得到[9]。以F1的合成为例,Schlenk管中加入3, 6二溴菲酮(1eq)、K2CO3(3eq)和4-硼酸三苯胺(3eq),然后抽真空充N2三次,加入无水异丙醇作为溶剂,接着加入在无氧环境下制备得到的PdCl2(0.05eq)和三苯基膦(0.15eq)配体的甲苯溶液,并注射入除氧的碳酸钠(3eq)水溶液,在80℃油浴下反应12h。粗产物通过快速硅胶柱除去不溶物,最终产物进一步通过硅胶色谱柱(CH2Cl2/石油醚体积比1:3)分离纯化得到。
3, 6-双(4-(二苯基氨基)苯基)-9H-芴-9-酮(F1):暗红色固体(产率72%)。1H NMR(400MHz,CDCl3)δ:7.77(s,2H),7.71(d,J=7.7Hz,2H),7.55(d,J=8.2Hz,4H),7.50(d,J=7.7Hz,2H),7.30(t,J=7.7Hz,8H),7.16(t,J=8.1Hz,12H),7.07(t,J=7.3Hz,4H);13C NMR(101MHz,CDCl3)δ:192.9,148.4,147.4,147.1,144.9,133.5,133.2,129.4,127.9,127.2,124.8,124.6,123.5,123.2,118.4;MS (EI) m/z:理论值C49H34N2O 666.3;实测值666.3 [M]+。
2, 7-双(4-(二苯基氨基)苯基)-9H-芴-9-酮(F2):红色固体(产率59%)。1H NMR (400MHz,CDCl3)δ:7.87(s,2H),7.67(d,J=7.6Hz,2H),7.57~7.48(m,6H),7.29~7.25(m,8H),7.13(d,J=7.8Hz,12H),7.05(t,J=7.3Hz,4H);13C NMR (101MHz,CDCl3)δ:193.9,147.8,147.5,142.6,141.5,135.3,133.4,132.6,129.4,127.4,124.7,123.5,123.2,122.4,120.7;MS (EI) m/z:理论值C49H34N2O 666.3;实测值666.3 [M]+。
双-(4′-二苯基氨基-联苯-4-基)-甲酮(F0):黄色固体(产率66%)。1H NMR (400MHz,CDCl3)δ:7.89(d,J=7.9Hz,4H),7.68(d,J=7.9Hz,4H),7.53(d,J=8.3Hz,4H),7.30~7.24(m,8H),7.15(d,J=8.0Hz,12H),7.05(t,J=7.3Hz,4H);13C NMR(101MHz,CDCl3):δ:195.8,148.1,147.5,144.6,136.0,133.3,130.8,129.5,128.0,126.3,124.8,123.4,124.8,123.3;MS (EI) m/z:理论值C49H36N2O 668.3;实测值668.3 [M]+。
3, 6-双(4-(二苯基氨基)苯基)菲-9, 10-二酮(P1):深红色固体(产率65%)。1H NMR (400MHz,CDCl3)δ:8.27~8.24(m,4H),7.66(d,J=8.2Hz,2H),7.59(d,J=8.4Hz,4H),7.31(t,J=7.6Hz,8H),7.17(d,J=8.0Hz,12H),7.10(t,J=7.3Hz,4H);13C NMR (101MHz,CDCl3)δ:179.9,149.0,148.0,147.2,136.2,132.3,131.2,129.5,129.4,128.1,127.4,125.1,123.8,122.8,121.7;MS (EI) m/z:理论值C50H34N2O2 694.3;实测值694.3[M]+。
2, 7-双(4-(二苯基氨基)苯基)菲-9, 10-二酮(P2):灰色固体(产率48%)。1H NMR (400MHz,CDCl3)δ:8.31(s,2H),7.95(d,J=8.4Hz,2H),7.85(d,J=8.4Hz,2H),7.51(d,J=8.4Hz,4H),7.28(t,J=7.6Hz,8H),7.13(d,J=8.0Hz,12H),7.06(t,J=7.4Hz,4H);MS(EI) m/z:理论值C50H34N2O2 694.3;实测值694.3[M]+。
2, 6-双(4-(二苯基氨基)苯基)蒽-9, 10-二酮(A1):红色固体(产率58%)。1H NMR (400MHz,CDCl3)δ:8.50(s,2H),8.33(d,J=8.0Hz,2H),7.95(d,J=8.0Hz,2H),7.61(d,J=8.3Hz,4H),7.32~7.28(m,8H),7.16(d,J=7.9Hz,12H),7.12~7.04(m,4H);13C NMR (101MHz,CDCl3) δ:183.0,148.8,147.3,146.3,134.1,132.0,131.7,131.3,129.5,128.1,128.0,125.0,124.7,123.6,122.9;MS (EI) m/z:理论值C50H34N2O2 694.3;实测值694.3 [M]+。
通过上转换荧光法测试了目标化合物在720~880 nm波段范围内的双光子激发荧光(2PEF)光谱,进而计算得到化合物的双光子吸收截面[10]。激发光源采用飞秒激光器(Mode-locked Tsunami Ti:sapphire Laser,80 MHz,710~880 nm,≤100fs)。脉冲峰宽小于100fs,大幅降低了测试样品的激发态吸收干扰。参比样品采用若丹明B的甲醇溶液(Φfl=0.50)[10]。
化合物的吸收光谱与发射光谱如图 1所示,主要的光谱数据列于表 1。吸收光谱中在长波长范围(350~600 nm)的吸收峰主要对应分子内电荷转移(ICT)吸收峰,在短波长范围(< 350nm)的吸收峰主要对应局域电子跃迁吸收峰。
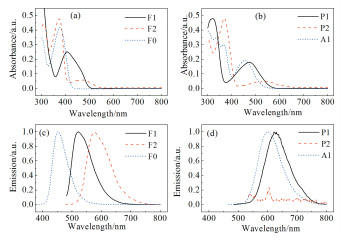
(a)芴酮系列吸收光谱;(b)芳香醌系列吸收光谱;(c)芴酮系列荧光发射光谱;(d)芳香醌系列荧光发射光谱
 下载:
导出CSV
下载:
导出CSV
| λabsmax /nma |
λemmax /nm b |
Δvss (103cm-1)c |
ε (104 mol· L-1·cm-1)d |
Φe | σ2 /GMf |
|
| F1 | 407 | 521 | 5.38 | 2.51 | 0.252 | 398 |
| F2 | 374 | 420 | 2.93 | 4.78 | 0.043 | 322 |
| 467 | 578 | 4.11 | 0.58 | |||
| F0 | 377 | 452 | 4.40 | 4.19 | 0.222 | 518 |
| P1 | 473 | 622 | 5.06 | 1.80 | 0.059 | 162 |
| P2 | 369 | 419 | 3.23 | 4.80 | (0.007) | |
| 535 | - | 0.48 | ||||
| A1 | 455 | 598 | 5.26 | 1.90 | 0.069 | 257 |
| (a)最大吸收峰位置;(b)最大发射峰位置;(c)Stokes位移;(d)摩尔吸光系数;(e)荧光量子产率;(f)双光子吸收截面 | ||||||
相比二苯甲酮作为中心电子受体的分子F0,芴酮作为中心电子受体的分子F1表现出更好的分子平面性与共轭特征,电荷转移吸收峰显著红移,说明化合物的电荷离域特征对其吸收光谱有着明显影响。当分子中心引入吸电子能力更强的菲酮后,相应化合物P1、P2的吸收峰进一步红移,说明中心基团的电子给受体性质对吸收光谱有着显著影响。通过对比F1与F2和P1与P2,相同中心基团但不同取代位点得到的化合物会导致显著不同的吸收光谱,其中F2和P2的电荷转移吸收峰相比F1和P1的吸收峰位置进一步红移,并且吸收强度显著降低。说明相比三苯胺基团在酮羰基的对位取代,三苯胺基团在酮羰基的间位取代会导致苯胺基团到羰基的电荷转移特征减弱,同时可能影响两端二苯胺基团与中心联苯结构之间的分子内的电荷离域特征。
化合物的荧光发射光谱与其吸收光谱的变化规律基本一致,不同共轭母核和不同取代位点显著影响了荧光强度和发射峰位置。相比中心基团为芴酮的化合物F1和F2,中心基团为蒽醌(A1)或菲醌(P1,P2)的化合物的荧光量子产率显著降低,其中P2在长波吸收峰535nm激发下无荧光, 这可能与分子中更强的吸电子基团以及羰基导致分子激发态非辐射失活途径的增加有关。
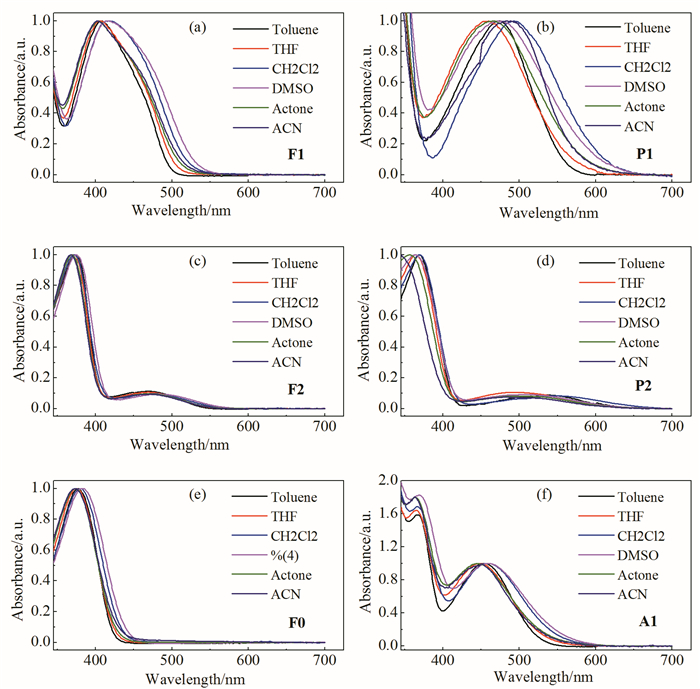
通过对比化合物在6种常见非质子溶剂(甲苯、四氢呋喃、二氯甲烷、二甲亚砜、丙酮、乙腈)中的紫外可见吸收光谱(图 2)与荧光发射光谱(图 3),可以看出随着溶剂极性的增大,6个化合物的吸收光谱无明显位移或位移幅度较小,而它们的发射光谱则大部分有着明显红移,导致化合物的Stokes位移显著增加。其中F0和F1的荧光发射峰红移相对更明显,而F2和P1的荧光发射峰红移相对较小,推测是由于F0和F1的电荷转移更显著,激发态极性更大。此外,化合物的电荷转移吸收峰均表现出溶剂猝灭现象,其中F2和A1在极性溶剂中的荧光猝灭现象非常明显,P2在所有溶剂中均没有荧光发射,这与电荷转移吸收峰特征相符。在F2和P2的定域吸收峰处激发得到了明显的荧光发射,其荧光强度随溶剂极性变化无明显猝灭现象,同时发射峰的溶剂位移效应并不显著,这与定域吸收π-π*跃迁特征相符。
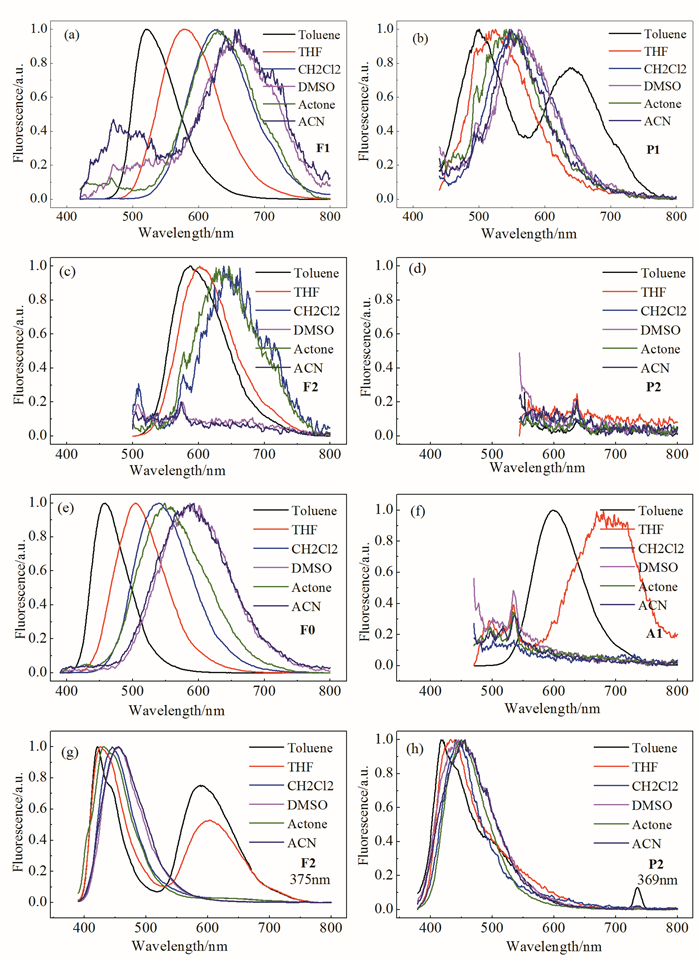
通过Lippert-Mataga公式分析溶剂极性与Stokes位移之间的关系,可以得到化合物的电荷转移特征[11, 12]:
|
${\nu _{\rm{A}}} - {\bar \nu _{\rm{E}}} = \frac{2}{{hc}}\left( {\frac{{\varepsilon - 1}}{{2\varepsilon + 1}} - \frac{{{n^2} - 1}}{{2{n^2} + 1}}} \right)\frac{{{{\left( {{\mu _{\rm{C}}} - {\mu _{\rm{E}}}} \right)}^2}}}{{{a^3}}} + const. $ |
其中,νA-νE代表Stokes位移,μG和μE分别代表基态和激发态偶极矩,ε代表溶剂介电常数,n代表折射系数,a代表Onsager空腔系数,h代表普朗克常数,c代表光速。
通过图 4分析可以发现,F1、F0和A1的Stokes位移较大,通过Lippert-Mataga方程拟合得到较大的斜率,说明跃迁过程中基态(S0)到激发态(S1)永久偶极矩变化较大,同时对应着明显的电荷转移过程。F2和P1的拟合直线对应的斜率较小,表明F2激发过程中的偶极矩变化较小,可能对应n-π*跃迁,这与吸收光谱中较弱的电荷转移吸收峰一致;比较意外的是,P1的偶极矩变化较小,这可能是由于化合物在基态存在电荷分离,共轭特征相对较弱所致。由于无荧光发射,P2的电荷转移吸收峰无对应的Stokes位移数据。但在F2和P2的定域吸收峰处激发所得到的Stokes位移也和溶剂表现出较好的线性关系,其较小的斜率可能与局域激发态性质有关。
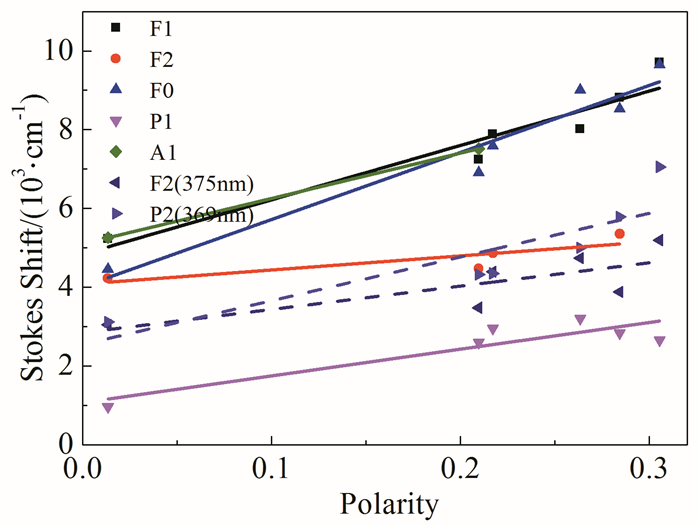
图 5显示了在不同比例THF/水混合溶液中化合物的荧光发射光谱。随着水的体积分数的上升,化合物分子逐渐聚集,化合物F1、F2和A1的荧光发光强度表现出先降低后增加的趋势。其中化合物F1在纯水环境中荧光显著强于A1和F2,在聚集条件下表现出了明显的聚集诱导荧光增强性质。可能原因是聚集状态显著影响了分子内以及分子间的能量传递过程,进而减少了激发态能量失活过程,从而导致荧光强度增加。
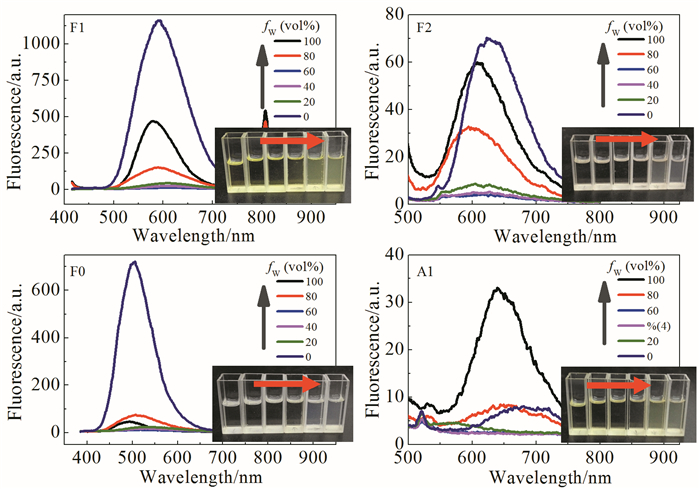
图 6显示了在甲苯中测试得到的化合物的双光子吸收光谱,参比为若丹明B的甲醇溶液(1.0×10-6mol/L)。通过对入射激光功率的对数值与化合物上转换荧光强度的对数值进行拟合,得到斜率接近2的线性关系,证明了其吸收机理为双光子吸收。
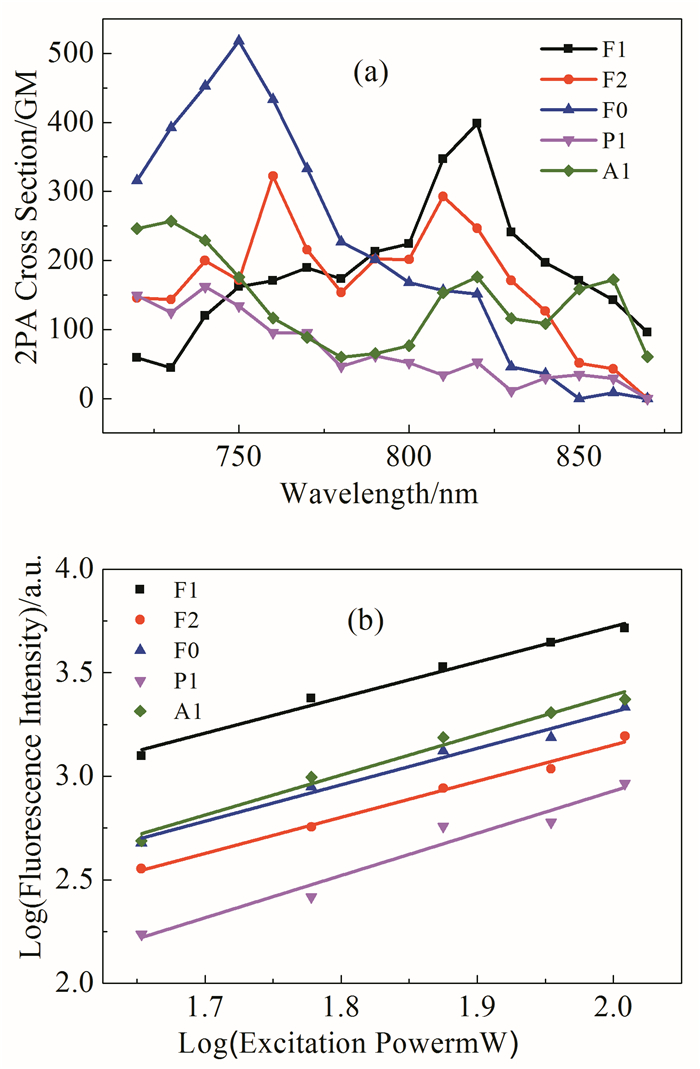
所有化合物均在720~880 nm范围表现出明显的双光子吸收,其中以芴酮为中心母核结构的化合物(F0,F1和F2)相比以菲醌(P1)和蒽醌(A1)为中心母核结构的化合物,表现出更大的双光子吸收截面,可能与芴酮结构化合物更好的电荷传输性质和共轭特性有关,这与由图 4得到的基态到激发态偶极矩变化特征趋势一致。基于二苯甲酮的F0和基于芴酮的F1由于荧光量子产率相对较大,表现出更好的双光子发光效率,在双光子生物荧光成像领域有着潜在的应用前景。
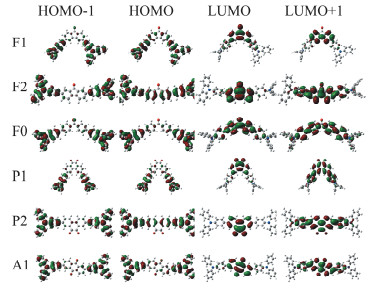
为了进一步理解不同结构特征的中心基团对分子光谱性质的影响,对化合物进行了基于密度泛函理论(DFT)的量化计算,计算在PBE0/6-311g(d)水平下进行[13]。所有目标化合物的前线分子轨道如图 7所示,其中二苯胺主要作为电子给体贡献给HOMO-1和HOMO,而苯酮、芴酮、蒽醌、菲醌等基团作为电子受体主要贡献给LUMO和LUMO+1。由于化合物的电荷转移吸收峰主要对应HOMO到LUMO的跃迁,通过对比可以发现,所有化合物都表现出了显著的电荷转移特征。芴酮作为中心母核结构的化合物F1和F2相比菲醌为中心母核结构的化合物P1和P2在HOMO轨道中的电荷离域特征更明显;同时苯环上羰基间位取代(F2和P2)相比对位取代(F1和P1)的分子在HOMO轨道中表现出更明显的电荷离域特征,说明中心共轭骨架对其共轭特性有着显著影响。这一系列计算结果与化合物的光谱变化特征是一致的。
本文设计合成了系列以芳香酮为母核结构的化合物,表征了其线性光物理性质与双光子吸收性质。通过对比化合物分子中心的共轭结构,从芴酮到菲醌和蒽醌,可以发现分子中心共轭结构显著影响分子的共轭性质和电荷转移性质。通过对比中心共轭基团相同但取代位点不同的的化合物,可以发现,对于芴酮和蒽醌结构,取代位点为羰基对位的化合物相比羰基间位的化合物表现出更明显的电荷转移特征。相比基于菲醌和蒽醌结构的化合物,基于芴酮结构的F1和基于二苯甲酮结构的F0表现出更好的共轭性质和电荷转移性质,得到了较大的双光子吸收截面以及较高的荧光量子产率,在生物荧光成像领域有着潜在的应用前景。
S Sasaki, G P C Drummen, G I Konishi. J. Mater. Chem. C, 2016, 4(14): 2731-2743. doi: 10.1039/C5TC03933A
A C Benniston, A Harriman. Chem. Soc. Rev., 2006, 35(2): 169-179. doi: 10.1039/B503169A
G Ramakrishna, T Goodson. J. Phys. Chem. A, 2007, 111(6): 993-1000. doi: 10.1021/jp064004n
G S He, L S Tan, Q Zheng et al. Chem. Rev., 2008, 108(4): 1245-1330. doi: 10.1021/cr050054x
F Helmchen, W Denk. Nat. Methods, 2005, 2(12): 932-940. doi: 10.1038/nmeth818
K S Lee, R H Kim, D Y Yang et al. Prog. Polym. Sci., 2008, 33(6): 631-681. doi: 10.1016/j.progpolymsci.2008.01.001
Z Sun, L P Zhang, F Wu et al. Adv. Funct. Mater., 2017, 27(48): 1704079. doi: 10.1002/adfm.201704079
K Rurack: Fluorescence Quantum Yields: Methods of Determination and Standards, Standardization and Quality Assurance in Fluorescence Measurements I, 2008: 101-145.
A L Capodilupo, V Vergaro, E Fabiano et al. J. Mater. Chem. B, 2015, 3(16): 3315-3323. doi: 10.1039/C4TB02116A
N S Makarov, M Drobizhev, A Rebane. Opt. Express, 2008, 16(6): 4029-4047. doi: 10.1364/OE.16.004029
D Cvejn, E Michail, K Seintis et al. RSC Adv., 2016, 6(16): 12819-12828. doi: 10.1039/C5RA25170B
J R Lakowicz. Principles of Fluorescence Spectroscopy. Springer Science & Business Media, 2013.
M J Frisch, G W Trucks, H B Schlegel et al. Gaussian 09. Wallingford, CT, 2009.

图 1 目标化合物的紫外可见吸收与荧光发射光谱(10-5mol/L甲苯溶液)
Figure 1 The UV-Vis absorption and fluorescence emissionspectra of target compounds (10-5mol/L in toluene)
(a)芴酮系列吸收光谱;(b)芳香醌系列吸收光谱;(c)芴酮系列荧光发射光谱;(d)芳香醌系列荧光发射光谱

图 2 化合物在不同极性溶剂中的吸收光谱
Figure 2 The UV-Vis absorption spectra of target compounds in different polar solvents

图 3 化合物在不同极性溶剂中的发射光谱(F2和P2的荧光发射光谱分别通过电荷转移吸收峰和局域吸收峰激发,其他化合物只用电荷转移吸收峰激发)
Figure 3 The fluorescence emission spectra of target compounds in different polar solvents (the spectra of F2 and P2 were obtained by ICT absorption peak and local absorption peak, others are only excited by ICT peak)

图 4 目标化合物的Stokes位移与溶剂极性的线性关系
Figure 4 The linear relationship between Stokes shift of target compounds and polarity of solvents

图 5 目标化合物的荧光发射光谱,溶剂为不同水体积分数(fw)的THF/水的混合溶液
Figure 5 Fluorescence emission spectra of target compounds in THF/water mixtures with different water volume fractions (fw)

图 6 (a) 目标化合物的双光子吸收光谱;(b)入射激光功率和上转换荧光的对数关系图
Figure 6 (a) 2PA spectra of target molecules; (b) Logarithmic relationship between excitation power and upconversion fluorescence intensity
表 1 化合物在甲苯中的光物理数据
Table 1. The photo-physical data of the compounds in toluene
| λabsmax /nma |
λemmax /nm b |
Δvss (103cm-1)c |
ε (104 mol· L-1·cm-1)d |
Φe | σ2 /GMf |
|
| F1 | 407 | 521 | 5.38 | 2.51 | 0.252 | 398 |
| F2 | 374 | 420 | 2.93 | 4.78 | 0.043 | 322 |
| 467 | 578 | 4.11 | 0.58 | |||
| F0 | 377 | 452 | 4.40 | 4.19 | 0.222 | 518 |
| P1 | 473 | 622 | 5.06 | 1.80 | 0.059 | 162 |
| P2 | 369 | 419 | 3.23 | 4.80 | (0.007) | |
| 535 | - | 0.48 | ||||
| A1 | 455 | 598 | 5.26 | 1.90 | 0.069 | 257 |
| (a)最大吸收峰位置;(b)最大发射峰位置;(c)Stokes位移;(d)摩尔吸光系数;(e)荧光量子产率;(f)双光子吸收截面 | ||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们