表 1
标题化合物的晶体学数据
Table 1.
Crystallographic data of the titled compound

香豆素是一种重要的天然产物,广泛存在于芸香科、伞形科、菊科、豆科、瑞香科、茄科等植物以及动物或微生物的代谢产物中[1]。香豆素是一种芳环内酯化合物,分子基本骨架为苯并吡喃酮。香豆素及其衍生物有很好的生物活性,具有抗肿瘤、抗HIV、抗炎等药理作用[2~5]。香豆素是电子离域的大共轭结构,其光吸收及荧光特性可随衍生基团的不同而发生很大变化。利用这一点,香豆素分子修饰不同的活性基团后,可作为荧光探针识别金属阳离子[6~8]、阴离子[9]、中性分子[10],甚至可区分出化学性质相似的不同稀土元素[11, 12]。其光致电子转移的特性还可作为染料敏化分子用于染料敏化太阳能电池[13, 14]。
密度泛函理论(DFT)是量子化学计算方法中性价比最高、应用最广泛的方法。在微观层面,用DFT解释和预测化学性质的做法已广泛应用于化学研究中,并且可获取传统实验及表征方法难以得到的重要信息。
对香豆素类化合物的研究主要集中于生物活性与荧光、光敏材料的实验探究中,但通过晶体和理论计算研究其弱相互作用与光致激发的还较少有报道,本文合成了一种香豆素衍生物,通过实验表征和理论计算相结合的手段研究其分子间弱相互作用、光致激发过程,并预测表面活性位点,为该分子以及该类化合物在生物医药、分子传感器、光电转化材料等领域的应用提供理论和实验数据的支持。
所用试剂均为市售分析纯,用前未做进一步纯化。Bruker AVANCE Ⅲ 600MHz超导核磁共振谱仪;Burker VERTEX 80V红外光谱仪;Lambda 950紫外可见光光谱仪;Nonius CAD4单晶衍射仪。
参照文献[15]的方法,在100mL圆底烧瓶中依次加入40mL无水乙醇、0.6mL 3-叔丁基水杨醛、0.3mL乙酰乙酸乙酯、5滴乙酸、5滴哌啶,加热回流5h。室温下冷却过夜,抽滤后得到淡黄色固体,粗产品用无水乙醇重结晶,得到白色固体0.36g(产率73.2%)。熔点158℃。1H NMR (600MHz,CDCl3) δ:1.46 (s,9H,-C(CH3)3),2.55(s,3H,-CH3),7.34~7.36 (m,1H,Ar-H),7.69 (d,J=7.2Hz,1H,Ar-H),7.82(d,J=7.2Hz,1H,Ar-H),8.654 (s,1H, C=CH-);13C NMR (151MHz,CDCl3) δ:29.8,30.7,35.0,118.8,123.5,124.6,128.6,132.1,138.1,148.5,154.1,158.9,195.7。
将3-乙酰基-8-叔丁基香豆素的白色固体溶于无水乙醇中,室温下自然挥发,两天后即可得到适合单晶测试的3-乙酰基-8-叔丁基香豆素的白色棒状晶体。选取大小合适的晶体,在Nonius CAD4单晶衍射仪上收集数据,采用经石墨单色化的MoKα(λ=0. 071073nm)的射线为光源,以Φ-ω方式在1.84°≤θ≤25.35°范围内扫描,293(2) K下共收集1227个衍射点,其中独立衍射点586个。结构经直接法用SHELXS-97解得,并用SHELXL-97进行校正。非氢原子的坐标及各向异性热参数用全矩阵最小二乘法进行最后修正,氢原子坐标由理论加氢后,再进行各向同性修正得到。结晶体数据采用Crystal Structure程序包(Rigaku NSC,2001)计算得到。标题化合物的晶体学参数列于表 1,主要键长和键角的计算值和实验值列于表 2。CCDC:1866825。
 下载:
导出CSV
下载:
导出CSV
| Formula | C15H16O3 |
| Formula weight | 244.28 |
| crystal system | Orthorhombic |
| a/Å | 8.3150(17) |
| b/Å | 6.9500(14) |
| c/Å | 22.153(4) |
| a/(°) | 90.00 |
| β/(°) | 90.00 |
| r/(°) | 90.00 |
| V/Å3 | 1280.2(4) |
| T/K | 293(2) |
| Z | 8 |
| 计算密度/g·cm-3 | 1.267 |
| F(000) | 520 |
| 衍射点(总数) | 1227 |
| 独立衍射点 | 586 |
| R1((I > 2σI)) | 0.1236 |
| wR2 | 0.0832 |
| 拟合度的优良度 | 1.002 |
| w=1/[σ2(F0)2+(0.0230P)2+0.0000P], where P=((F0)2+2Fc2)/3 | |
下载:
导出CSV
| Bond Length/Å | Bond Angle/(°) | |||||
| X-ray | B3LYP | X-ray | B3LYP | |||
| O(1)-C(8) | 1.378(3) | 1.365 | C(8)-O(1)-C(9) | 123.6(3) | 125.1 | |
| O(1)-C(9) | 1.389(4) | 1.399 | C(2)-C(1)-C(9) | 118.8(3) | 119.3 | |
| C(1)-C(2) | 1.350(4) | 1.364 | C(2)-C(1)-C(10) | 117.6(3) | 118.0 | |
| C(1)-C(9) | 1.438(4) | 1.463 | C(9)-C(1)-C(10) | 123.5(3) | 122.8 | |
| C(1)-C(10) | 1.509(5) | 1.508 | C(8)-C(3)-C(4) | 119.2.5(3) | 119.4 | |
| O(2)-C(9) | 1.201(4) | 1.210 | C(8)-C(3)-C(2) | 118.0(3) | 117.8 | |
| C(2)-C(3) | 1.419(4) | 1.429 | C(4)-C(3)-C(2) | 122.8(3) | 122.8 | |
| O(3)-C(10) | 1.203(4) | 1.223 | C(5)-C(4)-C(3) | 118.8(3) | 119.3 | |
| C(3)-C(8) | 1.383(4) | 1.411 | C(4)-C(5)-C(6) | 119.9(3) | 120.0 | |
| C(3)-C(4) | 1.402(4 | 1.409 | C(7)-C(6)-C(5) | 123.8(3) | 123.4 | |
| C(4)-C(5) | 1.376(5) | 1.381 | C(6)-C(7)-C(8) | 114.7(3) | 115.2 | |
| C(5)-C(6) | 1.387(4) | 1.405 | C(6)-C7)-C(12) | 121.8(3) | 122.2 | |
| C(6)-C(7) | 1.379(4) | 1.399 | C(8)-C(7)-C(12) | 123.5(3) | 122.5 | |
| C(7)-C(8) | 1.404(4) | 1.414 | O(1)-C(8)-C(3) | 119.2(3) | 119.1 | |
| C(7)-C(12) | 1.533(4) | 1.543 | O(1)-C(8)-C(7) | 117.3(3) | 118.3 | |
| C(10)-C(11) | 1.469(5) | 1.511 | C(3)-C(8)-C(7) | 123.5(3) | 122.6 | |
| C(12)-C(14) | 1.536(3) | 1.542 | O(2)-C(9)-O(1) | 115.3(3). | 116.2 | |
| C(12)-C(13) | 1.536(4) | 1.549 | O(2)-C(9)-C(1) | 127.6(4) | 127.9 | |
量子化学计算由Gaussian 09 D01程序包[16]完成,使用B3LYP[17]与M06-2X[18]DFT方法,结合D3(0)色散矫正[19]。计算晶体中分子间弱相互作用选取晶体结构中对应二聚体团簇,在B3LYP-D3/6-31+G**[20, 21]水平限制性优化(只优化H原子),再分别在B3LYP-D3/6-311+G**[22]与M06-2X-D3/6-311+G**水平计算二聚体结合能,并采用counterpoise方法校正基组重叠误差(BSSE),使用独立梯度模型IGM[23]将分子间弱相互作用图形化。计算红外光谱在B3LYP/6-31G*水平全原子优化,并在相同水平计算频率,所得频率乘以该水平下的频率校正因子0.9614 [24]以校正谐振近似误差,模拟红外光谱用Lorentz函数展宽,半峰宽设置为20cm-1(为了使理论计算和实际谱图相对应,需要根据相应函数进行人为展宽,展宽出的峰的积分面积与强度值成正比,对于分子振动光谱(对于电子光谱一般采用高斯函数进行展宽),一般采用洛伦茨函数进行展宽:
|
$L\left( x \right) = \frac{{{\rm{FWHM}}}}{{2\pi }} \cdot \frac{1}{{{x^2} + 0.25 \times {\rm{FWH}}{{\rm{M}}^2}}}$ |
其中,FWHM(Full width at half maximum,半高全宽)越大,展宽出的峰也越大,其是需要结合理论计算结果和实际测试谱图而人为给出的数据。
计算UV-Vis光谱在B3LYP/6-31G*[25]水平优化分子,并使用含时密度泛函理论(TD-DFT)[26]在TD-B3LYP/6-31G*水平结合SMD[27]溶剂化模型计算在二氯甲烷溶剂中激发能最低的30个单重激发态,模拟紫外可见光谱用Gaussian函数展宽,半峰宽设置为0.8eV。除限制性优化的二聚体团簇外,其余所有分子均在优化水平计算频率,确保其为能量极小点(没有虚频)。表面定量分析使用B3LYP/6-31G*水平计算的波函数,由Multiwfn[28]软件的定量分子表面分析功能[29]计算数据格点。电子跃迁的空穴-电子图,分子轨道等值面图,模拟红外、紫外可见光谱,分子表面图均由Multiwfn软件计算数据,并使用VMD[30]软件或Multiwfn软件绘制。
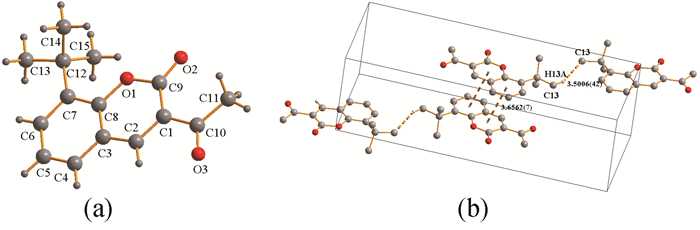
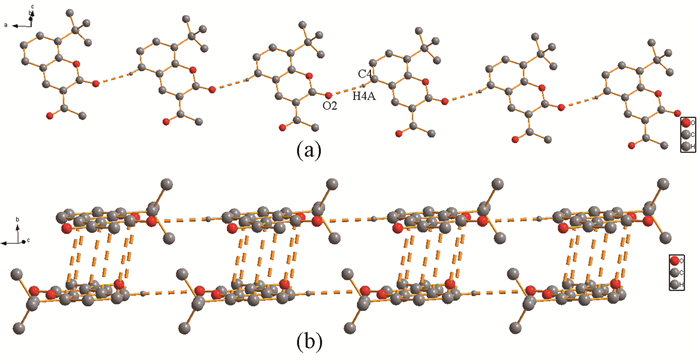
标题化合物的分子结构如图 1所示,它是一个中性分子,每个晶胞中含有4个3-乙酰基-8-叔丁基香豆素,它属于斜方晶系,空间群为Pnma。4个化合物分子之间通过弱的C(13)-H(13A)…C13i(H…C距离为3.500(6)Å,i: 2-x,0.5+y,1-z)氢键作用以及π…π相互作用(距离为3.538(4) Å)连接在一起。该化合物分子与简单香豆素分子一样,是一个完全共平面结构。测定的几何结构数据与理论计算所得数据接近(见表 2)。相邻分子与分子之间通过C(4)-H(4A)…O(2)ii(H…O距离为2.48Å,ii: 1+x,y,z)弱氢键相互作用形成一维链状结构,如图 2。链与链之间通过苯环与内酯环之间的C…C堆积作用(3.65~3.67Å)相互嵌入形成二维层状结构。
DFT计算也证实了晶体中这两种弱相互作用形式。由图 3的IGM等值面图可知,链中两分子间以苯环上氢和羰基氧形成的氢键为主,并存在叔丁基与芳环间范德华作用力,计算所得二聚结合能为3.9kcal/mol(M06-2X-D3)和4.6kcal/mol(B3LYP-D3);层间两分子以大环间的C…C堆积作用为主,并存在叔丁基与芳环的范德华作用力,计算所得二聚结合能为17.2kcal/mol (M06-2X-D3)和16.4kcal/mol (B3LYP-D3)。
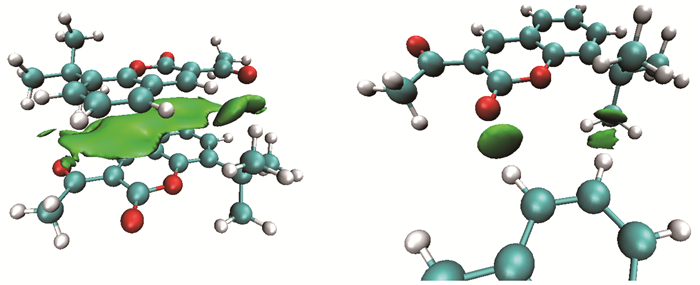
Green solid: weak interaction area; Isovalue: 0.007 a.u.; Level:B3LYP-D3/6-31+G**, restricted optimization in gas
图 4为标题化合物的红外光谱。上面曲线为实验光谱,下面曲线为B3LYP/6-31G*水平下模拟的理论光谱,振动类型通过理论计算所得简正振动模式指认,3500cm-1的宽峰在理论光谱中并无对应,应为水的氢键缔合峰。3072cm-1为苯环上C-H伸缩振动峰,2966cm-1为叔丁基C-H反对称伸缩振动峰,2875cm-1为叔丁基C-H对称伸缩振动峰,1737cm-1的强峰为内酯C=O伸缩振动峰,1596cm-1为苯环骨架伸缩振动峰,1358cm-1为乙酰基上C-H弯曲振动峰,1005和941cm-1均为大环整体骨架伸缩振动峰。745cm-1为大环骨架整体面外弯曲振动峰,555cm-1为大环骨架整体面内弯曲振动峰。除少量峰因谐振近似误差而吸收强度与实验值偏差较大,理论模拟光谱总体与实验光谱符合较好。
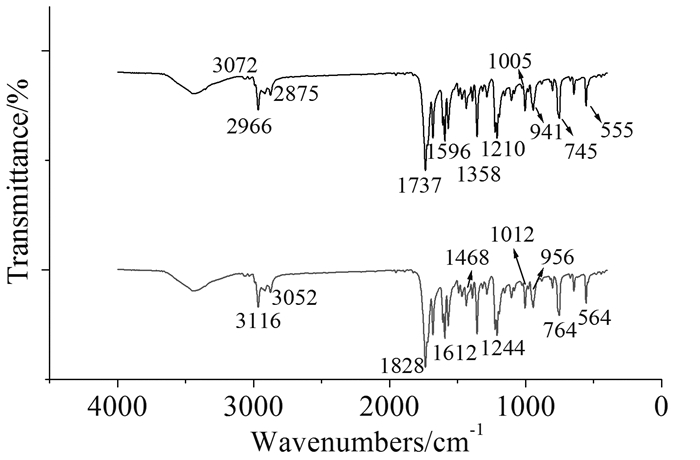
上:实验曲线;下:理论模拟曲线
标题化合物的实验光谱与理论模拟光谱见图 5,实验光谱在310nm处出现最大吸收,摩尔消光系数为16000L·mol-1·cm-1,并在350nm附近有一肩峰。理论计算表明,该分子在310nm处有一强吸收(振子强度f=0.3883),为电子基态到第3电子激发态的跃迁(S0→S3)。由图 5中空穴-电子图(跃迁产生正电空穴对应蓝色区域,跃迁产生富裕电子对应绿色区域)可知,该跃迁主要为苯环上π电子向酯环以及羰基氧上的跃迁,S0→S2跃迁吸收强度也较大(f=0.1287),主要也为苯环上π电子向酯环及羰基氧的跃迁,但分布与前者略有不同。S0→S3跃迁贡献了310nm处的吸收峰,S0→S2跃迁贡献了350nm附近的肩峰。S0→S1主要对应羰基氧上孤对电子向大环的跃迁,但振子强度为0,是一暗态。根据理论计算展宽所得模拟光谱与实验光谱在实验测定范围内符合较好,理论模拟光谱中190nm处还有一更大吸收峰,但超出实验测试范围。需要注意的是,紫外光谱的模拟是在基态优化的结构下计算基态到一批激发态的激发能和振子强度,然后把每个跃迁根据这两个量用高斯函数展宽,再把所有跃迁进行叠加,即得到UV-Vis光谱图。在本文的量化模拟中虽然考虑了溶剂效应,但实际体系中溶剂效应对激发能的影响是多方面的,会影响激发能而导致谱峰位移,还会影响吸收谱峰的强度,使谱峰加宽和变形,影响激发态结构等。

(Red curve: experiment spectrum (1.0×10-5mol/L in dichloromethane); Black curve: theoretical simulation spectrum; Black vertical line: normal mode; Blue area: isosurface of hole distribution of transition; Green area: isosurface of electron distribution of transition; Isovalue: 0.005 a.u.; Half band width: 0.8eV; Level: TD-B3LYP/6-31G* & SMD in dichloromethane.)
本文采用隐式溶剂模型,将溶剂环境当成具有一定介电常数的连续介质来考虑。这种方式考虑溶剂效应一般可以基本合理表现出它对激发能、振子强度的影响以及对激发态结构的影响,从图 5可以看出,理论模拟的UV-Vis和实际谱图符合的较好,而理论计算强度稍高于实际体系可能是由于实际体系中复杂的溶剂化效应所导致。
由图 6可知,分子表面正电区域分布于苯环边缘,羰基碳和烷烃基边缘,C(4)所连H原子附近出现整个分子中数值最大的21.79与21.64,所带正电最强,晶体结构中正是此处H与带显著负电的O(2)以静电作用结合形成了一维链上的氢键(晶体堆积还有规整性的要求),苯环上其余H所带正电也较多(20-21),在气相或溶液中可与带负电物质形成氢键等弱相互作用;酯基碳原子C(9)附近出现极值点15.74,可被亲核试剂进攻发生水解、氨解等反应,羰基碳原子C(10)附近极值点数值为11.43,虽带正电,但数值显著小于丙酮中的18.66,说明与内酯环中π电子形成共轭之后,羰基碳正电荷明显减小,更难在此处发生亲核加成反应。
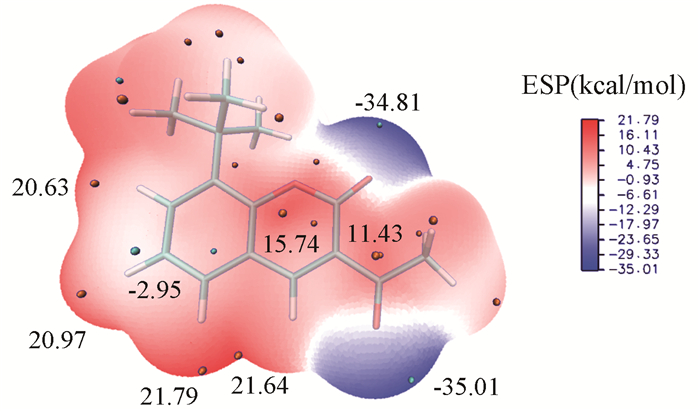
(Origin pellets: maximum point of ESP; Cyan pellets: minimum point of ESP; Numbers: value of maximum or minimum; Level: B3LYP/6-31G* in gas.)
由图 6可知,分子表面负电区域主要分布于酯基氧O(2)与羰基氧O(3)附近,且数值很负,分别为-34.81和-35.01,苯环上,尤其是C(5)上,也带有部分负电荷,数值为-2.95。O(2)与O(3)凭借显著的负电荷,可形成氢键或与金属阳离子配位形成配位键。苯环上的亲电取代反应最易发生于C(5)。苯环上方有一圈平均局部离子化能小的区域,该处电子活性高,为苯环上离域π电子,极小值点位于C(5)(9.56eV)和C(6)(9.74eV),且C(5)处为全局最小值点(图 7)。由静电势数值看,C(5)附近有静电势的极小值点(-2.95)而C(6)附近没有,说明亲电试剂在初期更易结合在C(5)附近形成弱相互作用复合物,而后与电子活性更高的C(5)形成共价键,且C(6)处在C(7)所连叔丁基的大空间位阻保护之下,使得亲电试剂更难进攻。综合静电,电子活性和空间位阻的因素,苯环上最易发生亲电反应的位置是C(5)。与苯及其衍生物横向比较,C(5)处发生亲电取代反应的活性小于苯(9.00eV),而介于氯苯的邻对位(邻位9.40eV,对位9.34eV)和硝基苯的间位(9.90eV)之间。内酯环上C(1)处电子活性也较高(9.84eV),也为可能的亲电反应位点,但附近并无静电势极小值点,说明此处不易以静电作用吸附亲电试剂,故反应活性不会太高[31]。
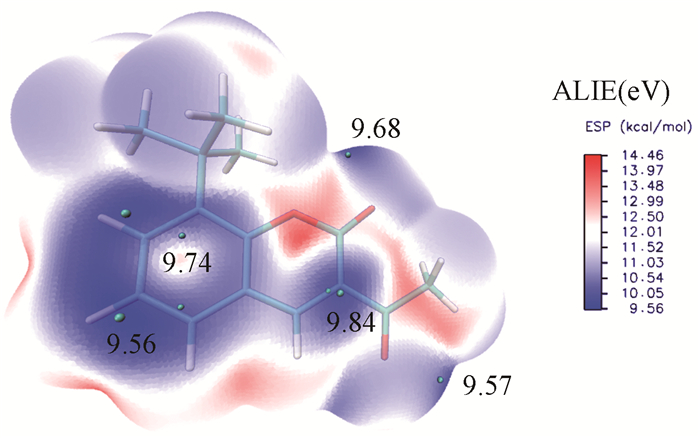
(Cyan pellets: minimum point of ALIE; Numbers: value of minimum point; Level: B3LYP/6-31G* in gas.)
本文用3-叔丁基水杨醛和乙酰乙酸乙酯合成了3-乙酰基-8-叔丁基香豆素,随后用X射线单晶衍射解析分子结构,并通过核磁共振谱、红外光谱、紫外可见光谱等方法表征,再用密度泛函理论方法进行量子化学计算,研究该化合物的晶体、谱学性质并预测其反应性质。结果表明,该分子是一个平面的大共轭分子,分子间存在一维方向延伸的氢键作用和层间π-π堆积作用,作用能分别为3.9和17.2 kcal/mol;近紫外、可见光范围内的最大吸收波长在310nm,摩尔消光系数为16000L·mol-1·cm-1,主要对应苯环上π电子向内酯环以及羰基氧上的跃迁;基于表面定量分析的理论研究预测该分子苯环上最易发生亲电反应的位置是酯基对位的6号碳原子,活性介于氯苯邻对位与硝基苯间位之间,羰基与酯基氧可形成氢键或与金属阳离子配位,酯基容易发生水解、氨解等反应,苯环上氢可形成氢键等弱作用。理论模拟的IR和UV-Vis谱图均与实验谱吻合,NMR、IR、UV-Vis与理论计算共同佐证了单晶衍射测定的分子结构的合理性。对分子表面反应位点,光致跃迁的研究可为该类化合物在生物医药、分子传感器、光电转化材料等领域的应用提供理论和实验数据的支持。
T C McKee, C D Covington, R W Fuller et al. Nat. Prod., 1998, 61(10):1252~1256. doi: 10.1021/np980140a
L M Bedoya, M Beltran, R Sancho et al. J. Bioorg. Med. Chem. Lett., 2005, 15(20):4447~4450. doi: 10.1016/j.bmcl.2005.07.041
L You, S Feng, R An et al. Nat. Prod. Commun., 2009, 4(2):297~302.
夏令先, 王玉斌, 黄文龙 等. 中国新药杂志, 2013, 22(20):2392~2404. http://www.cnki.com.cn/Article/CJFDTotal-ZXYZ201320014.htm
孔令雷, 胡金凤, 陈乃宏. 中国药理学通报, 2012, 28(2):165~168. http://www.cnki.com.cn/Article/CJFDTotal-YAOL201202006.htm
C Blackburn, M Q Bai, K A Lecompte et al. Tetrahed. Lett., 1994, 35(43):7915~7918. doi: 10.1016/0040-4039(94)80010-3
M Doludda, F Kastenholz, E Lewitzki et al. J. Fluoresc., 1996, 6(3):159~163. doi: 10.1007/BF00732055
E Brunet, M T Alonso, O Juannes et al. Tetrahed. Lett., 1997, 38(25):4459~4462. doi: 10.1016/S0040-4039(97)00901-5
马文辉, 彭孝军, 徐群 等. 化学进展, 2009, 19(9):1258~1266. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ200709004.htm
L Yuan, W Lin, Y Xie et al. J. Am. Chem. Soc., 2012, 134(2):1305~1315. doi: 10.1021/ja2100577
D Kovacs, X Lu, L S Meszaros et al. J. Am. Chem. Soc., 2017, 139(16):5756~5767. doi: 10.1021/jacs.6b11274
M Halim, M S Tremblay, S Jockusch et al. J. Am. Chem. Soc., 2007, 129(25):7704~7705. doi: 10.1021/ja071311d
韩亮, 周雪, 叶青. 有机化学, 2013, 33(5):1000~1004. http://www.cnki.com.cn/Article/CJFDTotal-YJHU201305014.htm
K D Seo, I T Choi, Y G Park et al. Dyes Pigm., 2012, 94(3):469~474. doi: 10.1016/j.dyepig.2012.02.015
D Lanari, R Ballini, A Palmieri et al. Eur. J. Org. Chem., 2011, 2011(15):2874~2884. doi: 10.1002/ejoc.201001476
M J Frisch, G W Trucks, H B Schlegel et al. Gaussian, Inc., Wallingford CT, 2013.
P J Stephens, F J Devlin, C F Chabalowski et al. J. Phys. Chem., 1994, 98(1~3):247~257.
Y Zhao, D G Truhlar. Theor. Chem. Acc., 2008, 119(5~6):525. doi: 10.1007/s00214-007-0401-8
S Grimme, J Antony, S Ehrlich et al. J. Chem. Phys., 2010, 132(15):154104. doi: 10.1063/1.3382344
W J Hehre, R Ditchfield, J A Pople. J. Chem. Phys., 1972, 56(5):2257~2261. doi: 10.1063/1.1677527
T Clark, J Chandrasekhar, G W Spitznagel et al. J. Comput. Chem., 1983, 4(3):294~301. doi: 10.1002/jcc.540040303
R Krishnan, J S Binkley, R Seeger et al. J. Chem. Phys., 1980, 72:650~654. doi: 10.1063/1.438955
C Lefebvre, G Rubez, H Khartabil et al. Phys. Chem. Chem. Phys., 2017, 19(27):17928~17936. doi: 10.1039/C7CP02110K
J P Merrick, D Moran, L Radom. J. Phys. Chem. A, 2007, 111(45):11683~11700. doi: 10.1021/jp073974n
P C Hariharan, J A Pople. Theoret. Chim. Acta, 1973, 28(3):213~222. doi: 10.1007/BF00533485
C Adamo, D Jacquemin. Chem. Soc. Rev., 2013, 42(3):845~856. doi: 10.1039/C2CS35394F
A V Marenich, C J Cramer, D G Truhlar. J. Phys. Chem. B, 2009, 113(18):6378~6396. doi: 10.1021/jp810292n
T Lu, F W Chen. J. Comput. Chem., 2012, 33(5):580~592. doi: 10.1002/jcc.22885
T Lu, F W Chen. J. Mol. Graph. Model., 2012, 38(9):314~323.
W Humphrey, A Dalke, K Schulten. J. Mol. Graph., 1996, 14(1):33~41. doi: 10.1016/0263-7855(96)00018-5
付蓉, 卢天, 陈飞武. 物理化学学报, 2014, 30(4):628~639. http://www.cnki.com.cn/Article/CJFDTotal-WLHX201404004.htm

图 1 (a) 标题化合物的分子结构图;(b)标题化合物的晶体结构图
Figure 1 (a)Molecular structure of the title compound; (b) The cell structure of the title compound

图 2 (a) 标题化合物通过C-H…O氢键作用形成的一维链状结构;(b)标题化合物通过C…C堆积作用形成的二维层状结构
Figure 2 (a) The 1D chain of the title compound through C-H…O hydrogen bonds; (b) The C…C stacking in the title compound

图 3 二聚体分子的独立梯度模型(IGM)等值面图
Figure 3 Isosurface map of Independent Gradient Model(IGM) of dimer molecule
Green solid: weak interaction area; Isovalue: 0.007 a.u.; Level:B3LYP-D3/6-31+G**, restricted optimization in gas

图 4 标题化合物的红外光谱图
Figure 4 IR spectrum of experiment and theoretical simulation of the title compound
上:实验曲线;下:理论模拟曲线

图 5 标题化合物的UV-Vis实验与理论模拟光谱
Figure 5 UV-Vis spectrum of experiment and theoretical simulation of the title compound
(Red curve: experiment spectrum (1.0×10-5mol/L in dichloromethane); Black curve: theoretical simulation spectrum; Black vertical line: normal mode; Blue area: isosurface of hole distribution of transition; Green area: isosurface of electron distribution of transition; Isovalue: 0.005 a.u.; Half band width: 0.8eV; Level: TD-B3LYP/6-31G* & SMD in dichloromethane.)

图 6 3-乙酰基-8-叔丁基香豆素表面静电势(ESP)填色图
Figure 6 Color-filled map of electrostatic surface potential(ESP) of the title compound
(Origin pellets: maximum point of ESP; Cyan pellets: minimum point of ESP; Numbers: value of maximum or minimum; Level: B3LYP/6-31G* in gas.)

图 7 标题化合物的表面平均局部离子化能(ALIE)填色图
Figure 7 Color-filled map of Average local ionization energy (ALIE) of the title compound
(Cyan pellets: minimum point of ALIE; Numbers: value of minimum point; Level: B3LYP/6-31G* in gas.)
表 1 标题化合物的晶体学数据
Table 1. Crystallographic data of the titled compound
| Formula | C15H16O3 |
| Formula weight | 244.28 |
| crystal system | Orthorhombic |
| a/Å | 8.3150(17) |
| b/Å | 6.9500(14) |
| c/Å | 22.153(4) |
| a/(°) | 90.00 |
| β/(°) | 90.00 |
| r/(°) | 90.00 |
| V/Å3 | 1280.2(4) |
| T/K | 293(2) |
| Z | 8 |
| 计算密度/g·cm-3 | 1.267 |
| F(000) | 520 |
| 衍射点(总数) | 1227 |
| 独立衍射点 | 586 |
| R1((I > 2σI)) | 0.1236 |
| wR2 | 0.0832 |
| 拟合度的优良度 | 1.002 |
| w=1/[σ2(F0)2+(0.0230P)2+0.0000P], where P=((F0)2+2Fc2)/3 | |
 下载: 导出CSV
下载: 导出CSV
表 2 标题化合物的键长键角的实验值和计算值
Table 2. Bond length and bond angle of the title compound (Level: B3LYP/6-31G*)
| Bond Length/Å | Bond Angle/(°) | |||||
| X-ray | B3LYP | X-ray | B3LYP | |||
| O(1)-C(8) | 1.378(3) | 1.365 | C(8)-O(1)-C(9) | 123.6(3) | 125.1 | |
| O(1)-C(9) | 1.389(4) | 1.399 | C(2)-C(1)-C(9) | 118.8(3) | 119.3 | |
| C(1)-C(2) | 1.350(4) | 1.364 | C(2)-C(1)-C(10) | 117.6(3) | 118.0 | |
| C(1)-C(9) | 1.438(4) | 1.463 | C(9)-C(1)-C(10) | 123.5(3) | 122.8 | |
| C(1)-C(10) | 1.509(5) | 1.508 | C(8)-C(3)-C(4) | 119.2.5(3) | 119.4 | |
| O(2)-C(9) | 1.201(4) | 1.210 | C(8)-C(3)-C(2) | 118.0(3) | 117.8 | |
| C(2)-C(3) | 1.419(4) | 1.429 | C(4)-C(3)-C(2) | 122.8(3) | 122.8 | |
| O(3)-C(10) | 1.203(4) | 1.223 | C(5)-C(4)-C(3) | 118.8(3) | 119.3 | |
| C(3)-C(8) | 1.383(4) | 1.411 | C(4)-C(5)-C(6) | 119.9(3) | 120.0 | |
| C(3)-C(4) | 1.402(4 | 1.409 | C(7)-C(6)-C(5) | 123.8(3) | 123.4 | |
| C(4)-C(5) | 1.376(5) | 1.381 | C(6)-C(7)-C(8) | 114.7(3) | 115.2 | |
| C(5)-C(6) | 1.387(4) | 1.405 | C(6)-C7)-C(12) | 121.8(3) | 122.2 | |
| C(6)-C(7) | 1.379(4) | 1.399 | C(8)-C(7)-C(12) | 123.5(3) | 122.5 | |
| C(7)-C(8) | 1.404(4) | 1.414 | O(1)-C(8)-C(3) | 119.2(3) | 119.1 | |
| C(7)-C(12) | 1.533(4) | 1.543 | O(1)-C(8)-C(7) | 117.3(3) | 118.3 | |
| C(10)-C(11) | 1.469(5) | 1.511 | C(3)-C(8)-C(7) | 123.5(3) | 122.6 | |
| C(12)-C(14) | 1.536(3) | 1.542 | O(2)-C(9)-O(1) | 115.3(3). | 116.2 | |
| C(12)-C(13) | 1.536(4) | 1.549 | O(2)-C(9)-C(1) | 127.6(4) | 127.9 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们