表 1
MNPs的合成方法及其性能
Table 1.
Synthesis methods and properties of MNPs

磁性纳米粒子(MNPs)代表了一类重要的功能性纳米粒子,因其有趣的纳米磁性和潜在的应用价值,使其得到了广泛研究。目前,在通过化学合成来精确控制MNPs尺寸方面已经取得重大进展,人们已经可以合成多种具有独特性能的MNPs,并能对其形状、组成、结构和多功能性进行调控[1]。这些研究不仅促进了人们对其独特纳米磁性的理解,也显示了这些MNPs在医学诊断和治疗[2, 3],尤其是磁共振成像(MRI)[4, 5]和磁流体热疗(MFH)[6]等方面的应用前景。
MNPs的一个最突出特征就是其磁性的尺寸依赖性。此外,对MNPs形状、组成和结构的精确控制也能够进一步调整它们的磁性。MNPs有着独特的尺寸效应。当一个MNP的尺寸低于某一临界尺寸时,其内部自由电子的自旋通过铁磁耦合被排列成一个方向,此时MNP成为单畴磁体。一个球形MNP达到单畴的临界直径(Dc)可以由式(1)估算:
|
$ {{D}_{c}}\approx \frac{36\sqrt{AK}}{{{\mu }_{0}}M_{s}^{2}} $ |
(1) |
其中,A是交换常数,K是衡量单体积内磁化方向翻转所需能量的有效各向异性常数,μ0是真空磁导率,Ms是饱和磁化强度[7]。
对于多畴MNPs,每个畴内磁化方向的取向由各向异性能KV(V是畴体积)和畴壁运动两者控制。在单畴MNPs中与此不同,不存在畴壁运动且磁化反转大部分依赖于KV。因此,单畴MNPs的Hc高于相应的多畴MNPs。然而,随着MNPs尺寸的不断下降,热能kBT(kB玻尔兹曼常数,T是绝对温度)开始与KV竞争(V是单畴NP的体积),导致Hc的下降[8]。当MNP的尺寸最终减小到临界值Ds,kBT超过KV,导致自发的磁性转化。在这个状态下,这些MNPs变为超顺磁性,它们不再具有矫顽磁性,但是仍然有着接近于它们相应的更大铁磁体的高的Ms。
MNPs的磁性性质还主要取决于它们的结构。晶体结构影响固有自旋-轨道相互作用,通常表现为磁晶各向异性。磁晶各向异性的大小由磁晶各向异性常数决定。通常,各向异性常数越大,矫顽力越大。MNPs也显示了形状依赖磁性。由于它们的各向同性结构,球形的MNPs没有形状各向异性。但是,当MNPs制成一维形状时,它们的易磁化轴可纵向对齐。理论计算表明,Fe MNPs的纵横比从1.1增加到1.5和2.0时,矫顽力分别提高了4倍和7倍[9]。形状各向异性增强的磁矫顽力已经在具有各种纵横比的Co纳米棒实验中被证明[10]。与超顺磁性Co MNPs相比,直径为15nm、长宽比为10的Co纳米棒在室温下的Hc为4500Oe[10]。形状各向异性效应对于FeCo合金纳米棒在高密度磁带记录中用作磁介质是非常重要的[11]。
在多组分MNPs体系中,MNPs的磁性可以通过改变组成进行调控。这在具有反型尖晶石结构的磁性铁氧体MFe2O4(M=Fe,Co,Ni和Mn)中可以看到[12]。在Fe3O4结构中,O离子组成了密堆积fcc结构,Fe离子位于八面体间隙(O)和四面体间隙(T)。Fe3O4可以写为[Fe3+]T[Fe2+Fe3+]OO4。在这种结构中,在T和O位中Fe3+的Fe-O-Fe键合导致了反铁磁性耦合和Fe3+磁矩的消除,因此,Fe3O4结构的总磁矩来自Fe2+的净磁矩。通过用Mn2+(d5)、Co2+(d7)或Ni2+(d8)代替Fe2+(d6),可以将MFe2O4的净磁化强度分别从4μB调整为5μB,3μB和2μB(μB,玻尔磁子,电子磁矩的自然单位)。相同的趋势也在一系列具有相同尺寸的12nm的单分散MFe2O4 MNPs中得到了验证[12],MnFe2O4、Fe3O4、CoFe2O4和NiFe2O4 MNPs的饱和磁矩分别为110、101、99和85 emu·gmetal-1。在结构中掺杂Zn2+可以进一步提高MFe2O4 MNPs的磁化强度[13]。MNPs磁化强度的这种组成调控对于进一步发展用于生物医学应用的灵敏磁探针十分重要。
本文讨论的MNPs主要是基于Fe、Co、Ni的铁磁元素、合金、氧化物或者复合结构,可以通过不同的化学方法制备得到。由于有机相合成对MNPs形成的控制很简单,并能保护这些MNPs对抗合成过程中的不可控氧化,因此有机相制备近年来比较受欢迎。这些典型的MNPs直径都小于20nm,通常是单磁畴,且表面原子比例呈指数增长,导致了它们磁性及其他性质的巨大改变。本文中我们简要介绍了MNPs的基本特征,接着介绍了一系列有着可控尺寸、形状、组成和结构的MNPs的有机相合成方法。最后,讨论了MNPs用于生物医学灵敏诊断与治疗中磁共振成像和磁流体热疗的最新进展。
MNPs可以通过溶胶-凝胶过程在水相溶液中制备,如共沉淀方法、微波合成法和水热反应;MNPs也能够在有机溶液反应中制备。有机相合成已用于制备基于Fe、Co和Ni的合金、氧化物、核/壳和哑铃结构的MNPs。MNPs的大小、形状和组成受一个或多个反应参数的影响,如反应物浓度、溶剂极性和反应温度/时间。在此,我们重点介绍了单分散MNPs的合成(表 1)。
 下载:
导出CSV
下载:
导出CSV
| 成分 | 反应试剂 | 表面活性剂 | 溶剂 | 形貌尺寸 | 磁性能 | 参考文献 |
| Fe | Fe(CO)5 | 油胺 | 1-十八烯 | 球形, < 10nm | Ms=102.6emu·g-1 | [16] |
| Fe(CO)5 | 油酸,油胺 | 二辛基醚 | 球形,5~20nm | Ms=173emu·g-1 | [20] | |
| Fe(CO)5 | 油胺,十六烷基氯化铵 | 1-十八烯 | 立方形,15nm | Ms=180emu·g-1, Hc=800Oe (5K) |
[21] | |
| Fe[N(SiMe3)2]2 | 油酸,十六烷基胺 | 均三甲苯 | 立方形,~6.2nm | Mr=83 emu·g-1 Ms=212 emu·g-1 |
[23] | |
| Fe[N(SiMe3)2]2 | 棕榈酸,十六烷基胺 | 均三甲苯 | 方形,13~20nm | - | 24 | |
| Co | Co2(CO)8 | 油酸,二正辛胺 | 二苯醚 | 核壳结构,球形,8~14nm | Ms=125 emu·g-1 | [26] |
| CoCl2(超氢化物) | 油酸,三烷基膦 | 二辛基醚 | 球形纳米晶,2~11nm | Hc=500 Oe (5K) | [30] | |
| Ni | NiAc2 | 油酸,三丁基膦 | 二苯醚 | 球形, < 6nm | Hc=3000~5000 Oe | [31] |
| Ni(acac)2 | 油胺,三正辛基膦 | 苄基醚 | 六角超晶格,4~12nm | - | [32] | |
| Ni(COD)2 | 十六烷基胺, 三正辛基氧膦 | 四氢呋喃 | 纳米棒,4×15nm | Ms=60 emu·g-1, Hc=275 Oe (2K) |
[33] | |
| Ni(acac)2 | 三正辛膦, 十六烷基胺 | 均三甲基苯 | 纳米立方体,12nm | Hc=105 Oe (10K), Mr=3.2 emu·g-1, Ms=12.4 emu·g-1 |
[34] | |
| FeCo | Fe(CO)5, Co2(CO)8 | 油酸,三辛基膦 | 1, 2-二氯苯 | 球形,1~11nm | Hc=17 Oe (室温) | [35] |
| Fe(CO)5 Co(N(SiMe3)2)2 | 油酸,十六烷基胺 | 甲苯 | 球形,15nm | Ms=160A·m2·kg-1 | [36] | |
| Fe(acac)3, Co(acac)2 | 油酸,油胺 | 油胺 | 球形,20nm | Ms=207 emu·g-1 Hc=300 Oe (室温) |
[37] | |
| FePt | Fe(CO)5,Pt(acac)2 | 油酸,油胺 | 二辛基醚 | 六角密堆积超晶格,3~10nm | Hc=180 Oe(5K) | [39] |
| Fe(CO)5, Pt(acac)2 | 油酸,油胺 | 苯醚 | 纳米立方体,3~9nm | Hc=22 kOe (室温) | [46] | |
| Na2Fe(CO)4, Pt(acac)2 | 油酸 | 十九烷 | 面心立方晶体,<3nm, | Hc=3100 Oe (10K) | [41] | |
| FeCl2, Pt(acac)2/ superhydride |
油酸,油胺 | 二苯醚 | 球形,4nm | Hc=2400 Oe (室温) | [42] | |
| CoPt3 | Co2(CO)8,Pt(acac)2 | 1-金刚烷甲酸,十六胺 | 二苯醚 | 球形纳米晶,1.5 ~7.2nm | - | [43] |
| Co2(CO)8,Pt(hfac)2 | 油酸 | 甲苯 | 球形,<10nm | Hc=6900 Oe(5K) | [44] | |
| Co2(CO)8,Pt(hfac)2 | 十二烷异腈 | 壬烷 | 球形核壳结构,4.7nm | Hc=660 Oe(5K) | [45] | |
| FePd | Fe(CO)5,Pd(acac)2 | 1-金刚烷羧酸,三烷基膦 | 油酸 | 球形,11~16nm | Hc=350 Oe(2K) | [47] |
| FePd-Fe3O4 | Fe(CO)5,Pd(acac)2 | 油酸,油胺 | 1-十八烯 | 海胆状,60nm | Hc=0.8~2.6 kOe, Ms=90~190 emu·g-1 |
[48] |
| FePd/Pd | Fe(CO)5,Pd(acac)2 | 油胺 | 四氢化萘 | 核壳结构, 面心四方, 8.5nm | Ms=169 emu·g-1, Hc=1.0 kOe(室温) |
[49] |
| SmCo5 | Co2(CO)8,Sm(acac)3 | 油酸 | 二辛基醚 | 面心立方晶体,9nm | Hc=50Oe(300K) | [50] |
| Co2(CO)8,Sm(acac)3 | 油酸,油胺, 十六烷二醇 | 二辛基醚 | 球形,6~8nm | Hc=2.2 kOe(5K) | [51] | |
| Co2(CO)8,Sm(acac)3 | 油酸,二正辛胺 | 四氢化萘 | 球形,32nm | Hc=8 kOe(室温), Mr=40~50 emu·g-1 |
[52] | |
| Co(acac)2,Sm(acac)3 | 油酸, 三辛膦, 十六烷二醇 | 油酸 | 球形,25nm | Hc=12 kOe(室温), Ms=70 emu·g-1 |
[53] | |
| Fe3O4 | Fe(acac)3 | 油胺 | 苯醚 | 球形,7~10nm | Ms=80 emu·g-1 | [19] |
| Fe(acac)3 | 油胺,油酸 | 二苄醚 | 球形,3~20nm | Ms=82 emu·g-1 | [54] | |
| Fe3-xO4 | Fe(acac)3 | 癸酸 | 二苄醚 | 立方体,30nm | Ms=80~83 emu·g-1 | [57] |
| FePt-Fe3O4 | Pt(acac)2, Fe(CO)5, Fe(acac)3 | 油酸,油胺 | 辛基醚、二苯醚 | 核壳结构, 核4nm, 壳0.5~3nm | Hc=5.5 kOe(10K) | [63] |
| MnFe2O4/Fe | Fe(CO)5, Fe(acac)3, Mn2(CO)10 | 油酸,油胺 | 1-十八烯 | 核壳结构, < 8nm | Ms=149emu·g-1 | [68] |
| Fe3O4/Au/Fe3O4 | Fe(CO)5, HAuCl4 | 油酸,油胺 | 1-十八烯 | 哑铃型,Au 8nm, Fe3O4 20nm | Ms=80 emu·g-1 | [76] |
目前,单分散MNPs的形成机理尚未完全明确。根据LaMer和Dinegar提出的经典爆裂成核和生长模型,单分散MNPs的合成涉及几个连续阶段(图 1(a))[14]。在合成溶液中,当“单体”浓度增加到成核阈值(阶段Ⅰ)时,核开始形成;在阶段Ⅱ,发生“爆裂成核”,即大量成核,同时单体浓度迅速降低;当浓度下降到阶段Ⅲ的成核阈值以下时,成核终止,反应处于生长阶段,系统自由能驱动单体附着到预先形成的核上。生长过程一直持续直到单体浓度下降到饱和水平以下。为了合成单分散MNPs,缩短成核过程并保持每个核周围的均匀生长速度是非常重要的。
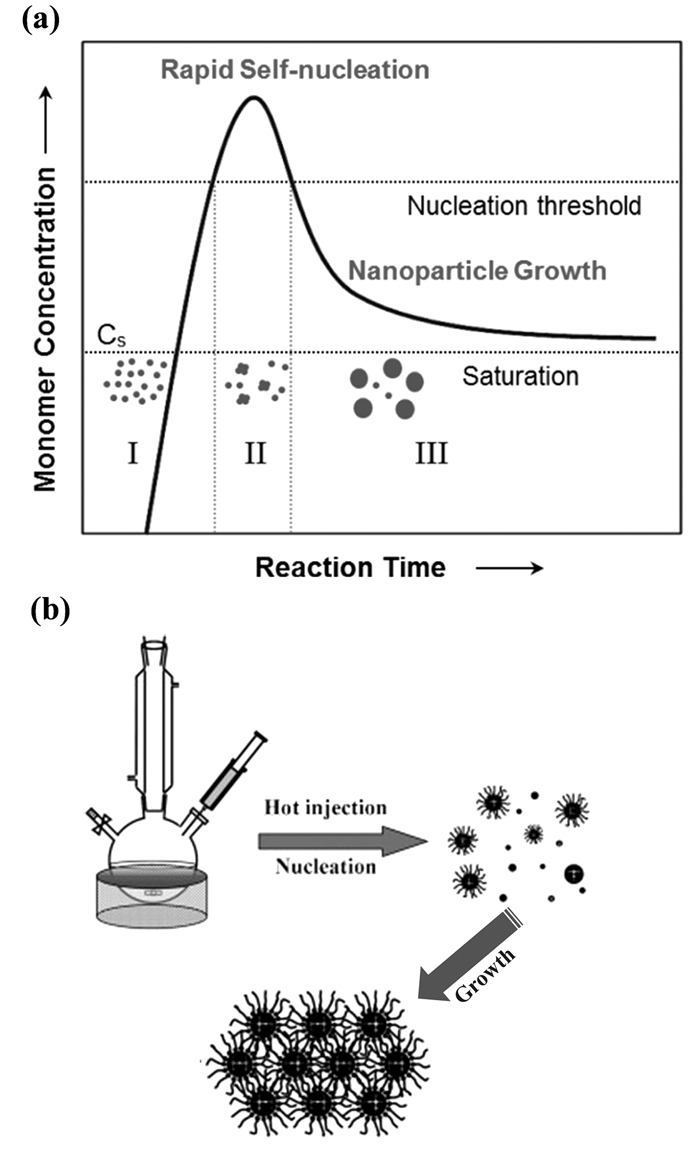
有两种方法可以达到有机相合成中的有效“爆裂成核”。一种方法是在单分散CdE(E=S,Se和Te)NPs的合成中引入“热注入”[15]。在预设高温下快速注入反应前驱体可导致CdE的爆发性成核,随后伴随着可控生长形成单分散CdE NPs,详见图 1(b)。这种热注射方法已经用于许多MNPs的合成,包括单分散的Fe[16]、Co[17]、Ni MNPs[18]。第二种是采用“升温”法,即前驱体、表面活性剂和溶剂在室温下混合,然后将混合物加热至所需温度以引发成核过程并控制MNPs生长[19]。与“热注入”法相比,“升温”法更适合于更复杂的MNPs(如核/壳和哑铃MNPs)的合成,因为这种复合结构的形成通常需要在预先种下的单分散MNPs上进行不均匀成核和第二组分的生长。
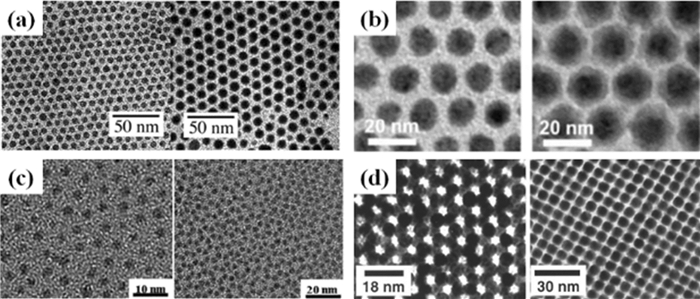
Fe是一种重要的铁磁材料,其质量磁化强度值约为220emu·g-1。可以通过Fe(CO)5的热分解来合成Fe MNPs。在研究的早期阶段,分解过程在聚合物基质中进行以防止形成的MNPs聚集。单分散Fe MNPs的需求促进了在有机溶液中热分解Fe(CO)5的合成研究。Farrell等[20]使用油酸和油胺作为表面活性剂,在二辛基醚中分解Fe(CO)5制备出了具有可控大小、尺寸在5~20 nm的单分散Fe MNPs(图 2(a))。但是这种方法制备的Fe MNPs暴露在空气中容易被氧化成氧化铁。为了提高Fe MNPs的抗氧化性,Peng等[16]通过控制三甲胺氮氧化物(Me3NO)的量将合成的Fe MNPs氧化成了核/壳Fe/Fe3O4 MNPs。具有Fe3O4结晶层的核/壳Fe/Fe3O4 MNPs显示出了在周围环境中抗空气氧化的稳定性。结晶度高的Fe MNPs化学稳定性更好。Lacroix等[21]发现,在氯化十六胺(HDA·HCl)存在下,Fe(CO)5的分解可以合成出晶态体心立方(bcc)Fe MNPs。HDA·HCl中Cl-的存在有利于晶态Fe MNPs的热力学生长。这些bcc-Fe MNPs比在没有HDA·HCl的情况下制备的非晶Fe MNPs磁矩更高且化学稳定性更好。
Fe(CO)5作为Fe和C源,通过预先制备的Fe MNPs和Fe(CO)5的反应可以制备核/壳Fe/FexC MNPs[22]。该FexC外壳在金属Fe核上起到有效保护层的作用,并且Fe/FexC MNPs具有比Fe/Fe3O4 MNPs更高的质量磁化值。此外,Dumestre等设计出了一种新的Fe复合Fe[N(SiMe3)2]2作为前驱体用来制备Fe MNPs[23]。在150℃的H2气氛中,HDA和油酸存在下,Fe[N(SiMe3)2]2的还原分解产生具有立方形状的高质量7nm bcc Fe NPs。使用棕榈酸代替油酸,在不同的HDA/棕榈酸比下,可合成出13~30 nm的尺寸可控的均匀Fe MNPs[24]。
块状的Co通常采用面心立方(fcc)或六角密堆积(hcp)结构。在二苯醚中使用油酸和三丁基膦(TBP)作为表面活性剂,热分解Co2(CO)8可以得到具有多级fcc结构的单分散Co MNPs[25]。Peng等[26]使用二正辛胺取代TBP,通过相同的分解反应制备得到了单分散的多晶Co MNPs(图 2(b))。Petit等[27]在反相胶束中通过NaBH4还原[Co-(AOT)2]制备得到了多晶Co MNPs。通过在二辛基醚中的溶液相退火,这些多晶Co MNPs可以转变为hcp-Co MNPs[28]。除了fcc和hcp结构,Co MNPs还能采取一种亚稳态的ε-结构[29]。单分散的ε-Co MNPs是在200℃时,油酸和三烷基膦存在下在二辛醚中用超氢化物(LiBEt3H,Et=乙基)还原CoCl2得到的[30]。通过TBP的空间效应,Co MNPs的尺寸控制在2~11 nm。亚稳态的ε-Co MNPs在300℃下可以转化为更稳定的hcp-Co MNPs,500℃下转化为fcc Co MNPs,其磁性也发生从软磁性ε-Co到硬磁性hcp-Co再到软磁性fcc-Co的改变[30]。
用于合成Ni MNPs的Ni前驱体非常有限,Ni(CO)4是著名的羰基化合物且热力学很稳定,但是其极高的毒性限制了它用作合成NPs的前驱体。合成Ni MNPs的常用Ni盐有NiAc2 (Ac=acetate),Ni(acac)2 (acac=acetylacetonate),Ni(oleate)2或Ni(COD)2 (COD=cycloocta-1, 5-diene)。通常选用NiAc2,在250℃下,以油酸、TBP和三丁胺(TBA)为表面活性剂,于二苯醚中由多元醇还原来合成Ni MNPs[31]。Ni MNPs的大小可以通过表面活性剂的体积来控制。使用硼烷三丁胺对Ni(acac)2的还原可以制备出超小的(3nm)Ni MNPs,图 2(c)是该方法制备得到的Ni MNPs的透射电镜(TEM)图。单分散Ni MNPs也可以通过将Ni(acac)2的油胺溶液“热注入”膦溶剂来制备[18]。替代“热注射法”,Cargnello等[32]将Ni(acac)2与油胺和三辛基膦(TOP)在苄基醚中混合,并以40℃/min的加热速率将温度从100℃加热至230℃也制备得到了单分散Ni MNPs。当使用Ni(COD)2作为前驱体、HDA作为表面活性剂时,就形成了Ni纳米棒[33]。一般认为表面活性剂HDA能够指导Ni在反应溶液中的各向异性生长。在HDA和TOP存在时,在1bar H2气氛下Ni(acac)2的还原产生12nm的Ni立方体[34]。TOP的量对立方体的形成很关键,过多的TOP会使产物变成球形的5nm MNPs。
众所周知,磁性元件中FeCo合金具有最高的磁矩,Ms可以达到245emu·g-1。合成FeCo MNPs的最佳金属前驱体就是Fe和Co的羰基化合物,如Fe(CO)5和Co2(CO)8在1, 2-二氯苯中同时热分解可合成出单分散的FeCo MNPs[35]。然而,Fe(CO)5的分解速率较慢,很难控制最终FeCo MNPs的组成。为了克服这一问题,Desvaux等[36]研发出了新的Co前驱体Co(η3-C8H13)(η4-C8H12)和Co(N-(SiMe3)2)2,其可与Fe(CO)5反应制备FeCo MNPs,在这种情况下的Fe/Co比例更容易控制。当这些FeCo MNPs在500℃的Ar中煅烧30min后,每个FeCo NP都覆盖有一层薄的碳外壳,有效保护了FeCo MNPs不被快速氧化。金属盐的还原也能被用于合成单分散FeCo MNPs。Chaubey等[37]在Ar+7% H2气氛下,于表面活性剂与还原剂1, 2-十六烷二醇混合物中通过Fe(acac)3和Co(acac)2的还原分解制备得到了均一的FeCo MNPs,通过改变金属前驱体的比例来控制Fe/Co的比例。单分散FeCo MNPs也可以通过核/壳Co/Fe MNPs的受控金属扩散来制备[38]。核/壳Co/Fe MNPs是通过将Co MNPs涂覆一层Fe而合成得到的,通过控制Fe涂层的厚度,最终FeCo MNPs的Fe/Co组成很容易控制。
作为铁磁性MNPs,由于其独特的结构依赖性磁性,MPt (M=Fe,Co)合金MNPs已经被广泛用于磁记录、永磁体和其他磁性应用等。合金FePt MNPs的合成可在二辛醚溶剂中的Fe(CO)5和Pt(acac)2的高温热分解反应来完成,FePt的组成由Fe(CO)5/Pt(acac)2的摩尔比控制[39]。该反应可合成出单分散的4nm FePt MNPs,通过种子介导生长法可进一步扩大FePt MNPs的尺寸,因此可制备得到具有可控尺寸的一系列FePt MNPs。Chen等[40]进一步研究表明,可通过简单的一步反应制备粒径可达10nm的FePt MNPs。图 2(d)显示的是他们制备得到的具有代表性的6nm FePt MNPs的TEM图像。
除了Fe(CO)5以外,其他羰基化合物如Na2Fe(CO)4也可与Pt(acac)2反应制备FePt MNPs,其Fe/Pt组成更易控制[41]。FePt MNPs的另一种合成方法是金属盐在非水解溶剂中的共还原作用。例如,用过酸酐同时还原二苯醚中的FeCl2和Pt(acac)2产生单分散的4nm FePt MNPs[42]。金属前驱体的初始摩尔比也保留在最终的FePt MNPs中。
用于合成FePt MNPs的方法也可扩展至CoPt MNPs的合成。在二苯醚中,Co2(CO)8热降解和Pt(acac)2还原可以制备得到单分散的CoPt3 MNPs,其尺寸在1.5~7.2 nm之间[43]。CoPt MNPs也可以通过Co2(CO)8和Pt(hfac)2 (hfac=六氟乙酰丙酮)之间的氧化还原转移金属法合成[44]。该法已用于Co MNP和Pt(hfac)2之间的反应来合成核/壳Co/Pt MNPs[45]。合金MPt MNPs的形状控制通常在特定的受控反应环境中实现。例如,通过控制油酸和油胺的加入顺序和Fe/Pt前驱体比例来合成立方FePt MNPs[46]。
FePd和FePt结构上相似,都可以采取化学无序的A1结构或者有序的L10结构。在1-金刚烷羧酸和三烷基膦存在下,Hou等[47]利用Fe(CO)5的热分解和Pd(acac)2还原制备得到了16nm的Al-FePd MNPs;当该反应在油酸和油胺的存在下进行时,得到海胆状FePd-Fe3O4纳米复合材料。Fe/Pd摩尔比由反应混合物中加入Fe(CO)5的量和反应温度来控制[48]。在500℃还原退火时,A1-FePd-Fe3O4纳米复合物转化为交换耦合的L10-FePd-Fe。部分有序的L10-FePd MNPs也可以通过在600℃下还原退火Pd/Fe3O4 MNPs来制备[49]。
SmCo5合金有着大的磁晶各向异性常数(K=~2×107J·m-3)和高的居里温度(Tc=~730℃),在高温永磁体应用中十分重要。早期SmCo5 MNPs的合成是在二辛基醚中由Co2(CO)8的分解和Sm(acac)3还原制备得到的,由此合成出了低Sm含量的MNPs,300K下其Hc仅为50Oe[50]。使用多元醇作为还原剂可合成出尺寸小于10nm的SmCo MNPs,5K下Hc达到了2.2kOe[51]。制备SmCo MNPs的另一种策略是用强还原剂还原SmCo氧化物MNPs。Hou等使用两步法在Ca存在下通过对核/壳Co/Sm2O3 MNPs进行高温(900℃)退火还原制备得到了SmCo5纳米晶[52]。通过调节Sm2O3壳的厚度,在还原退火后可得到不同纳米结构的SmCo,室温下它们的Hc达到了8kOe。类似地,Chaubey等[53]通过高温还原Sm2O3和Co MNPs制备出纳米结构的SmCo5 (或Sm2Co17)粒子。这些SmCo MNPs具有强铁磁性,室温下Hc达到12kOe,Ms达到70emu·g-1。
氧化铁MNPs,特别是立方和六方铁氧体,由于其磁性能和强大的化学稳定性而显示出许多重要应用。常见的立方铁氧体有Fe3O4、Fe2O3和MFe2O4(M=Co,Mn,Ni,Zn等),六方铁氧体有MO·6Fe2O3或MFe12O19(M=Ba,Sr等)。立方铁氧体在结构上是各向同性的,且磁性较软(较小的K和Hc),而六方晶体铁氧体磁性要硬得多。
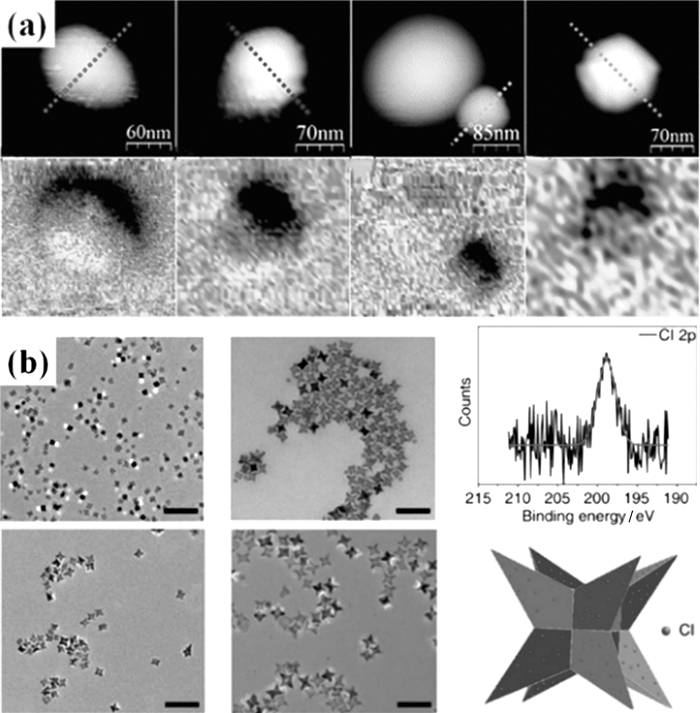
265℃下,Fe(acac)3在苯醚中热还原分解可得到单分散的Fe3O4 MNPs[19]。Sun等[54]把该反应的溶剂换为二苄醚,在300℃下制备得到了具有更好结晶度的6nm Fe3O4 MNPs。通过种子介导生长法,以4nm或6nm的MNPs作为种子,Fe3O4 MNPs的尺寸可以进一步增加到20nm。这种热分解方法可以容易地扩展用于制备MFe2O4 (M=Fe,Co,Ni,Mn和Zn),通过简单地在反应混合物中加入M(acac)2(也可以是MCl2)[54],用前驱体浓度[55]或仅使用油胺作为还原剂和表面活性剂[56]来控制尺寸。Moya等[57]在有机溶剂中用癸酸作为封端配体,通过Fe(Ⅲ)-乙酰丙酮化物的高温分解制备出了尺寸在25~30 nm的Fe3-xO4 NPs,并使用磁力显微镜(MFM)对单个纳米粒子的磁畴结构进行了表征并提出了单个纳米粒子的磁性反转机制。图 3(a)是所制备得到的纳米粒子及其MFM图。
合成氧化铁MNPs时可以使用Fe-油酸盐络合物作为起始前驱体,油酸盐铁络合物在三甲胺N-氧化物存在下可以热分解为单分散铁氧化物MNPs[58]。使用不同有机溶剂在不同回流温度下将铁-油酸盐络合物热分解可以产生均匀的氧化铁MNPs,其尺寸为5~22 nm。金属-油酸盐分解方法也可用于合成MFe2O4 MNPs(M=Co,Ni和Mn)[59]。
Zhao等[60]通过引入Cl-成功地制备出了尺寸可控的八足体氧化铁MNPs(图 3(b)),通过控制氧化铁MNPs的形态来实现高的横向弛豫率。八足铁氧化铁MNPs表现出超高的横向弛豫率值(679.3±30 L·mmol-1·s-1),表明这些八足体氧化铁可用作体内成像的T2造影剂。
寻找更先进的磁性纳米材料用于重要的技术应用需要制备出多功能的MNPs,并且能够将一定的附加功能与MNPs进行结合。这里我们重点关注了多功能磁性核/壳和哑铃状MNPs的有机相合成。通常它们通过类似于种子介导的生长过程制备得到,其中预合成的MNPs用作其他无机组分成核和生长的种子。
由于其独特的异质结构,有机相合成的核/壳MNPs已经得到了广泛研究。例如,用金属MNPs作为种子与O2或硒反应可合成出Fe/Fe3O4[16]、Ni/NiO[61]、Co/CoSe[62]等。当存在过量的O2、硒或TOP时,所有的MNPs都会发生反应,通过Kirkendall效应形成相应的空心氧化物、硒化物或磷化物。如合成Co/Au、Co/Pd、Co/Pt和Co/Cu MNPs所证明的,氧化还原转移法也用于合成核/壳MNPs[45]。
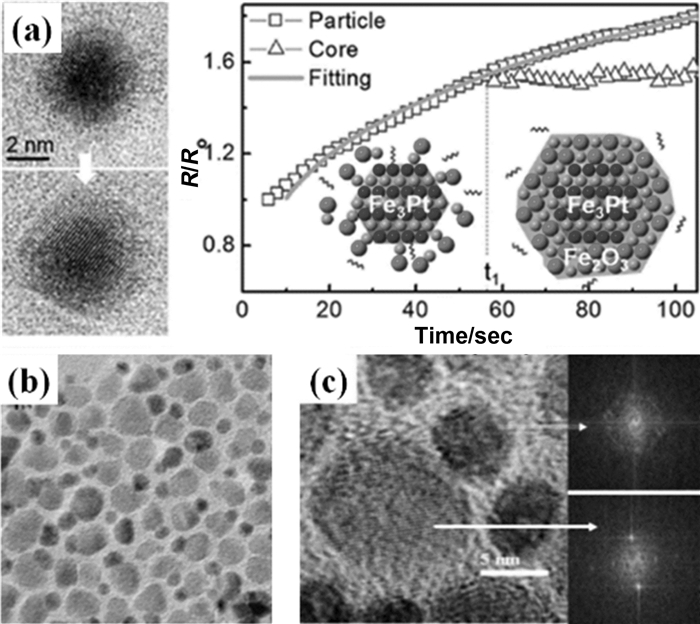
除了这些可控的氧化还原方法,合成磁性核/壳MNPs的更常见方法是种子介导生长法,即在已制备的MNPs核上生长均匀的壳。例如,在4nm FePt MNPs种子存在的条件下通过Fe(acac)3热分解可制备得到单分散的FePt/Fe3O4 MNPs[63]。通过控制FePt MNPs和Fe(acac)3的量,壳的厚度是可调节的。种子介导生长法也能用于在FePt MNPs核上涂覆其他铁氧体外壳[64]或软磁性外壳(如Co,Ni和Fe2C)[65],以及合成其他许多双磁性核/壳MNPs,包括CoFe2O4/MnFe2O4[66]、Zn0.4Fe2.6O4/CoFe2O4[67]、Fe/MFe2O4(M=Fe,Co和Mn)[68] MNPs。种子介导生长法能够被进一步拓展用于制备核壳结构的NPs如FePt/ZnO[69]、Fe3O4/ZnO[70]、Co/CdSe[71]、FePt/PbS[72]和Fe3O4/Cu2xS MNPs[73]。类似地,Au/Fe MNPs在空气中氧化得到核/空心壳Au/Fe3O4 MNPs[74],FePt/Co硫化(通过和S反应)得到核/空心壳FePt/CoS2 MNPs[75]。Au/Fe3O4 MNPs表现出氧化铁壳的超顺磁特性和Au核的等离子体特性[74],Fe3O4壳在Au核上的受控的各向同性成核和生长导致核/壳Au/Fe3O4 MNPs的形成,核与壳之间接触紧密[76]。Liang等[77]制备出了Fe3Pt/Fe2O3核-壳NPs(图 4(a))并使用原位液体池透射电镜对其生长进行了原位研究。通过控制前驱体溶液中Fe与Pt的比例来控制纳米粒子的生长,首先是Fe-Pt合金核的形成,随后是电子束诱导反应中氧化铁壳的形成。Fe-Pt合金核停止生长后,氧化铁壳的生长动力学没有实质性变化。他们的研究表明,Pt原子催化Fe离子的还原形成Fe3Pt合金核,当Pt耗尽时,氧化铁的直接沉淀导致核-壳纳米结构的形成。
不同于壳在种子MNPs上均匀形成得到的核/壳MNPs,哑铃型MNPs是在种子MNPs表面上的一种或更多离散组分的各向异性成核和生长而形成。非均相哑铃型MNPs的成功合成依赖于许多因素,包括不同组分之间的晶格匹配以及合成条件(例如溶剂极性、种子-前驱体比率、表面活性剂、生长速率和温度)。
研究最多的哑铃型MNPs是贵金属-磁性氧化物MNPs。哑铃型Au-Fe3O4 MNPs是通过Fe(CO)5在预先形成的Au MNPs表面的热分解制备的[78]。通过调节Fe(CO)5和Au种子的比例,Fe3O4 MNPs的尺寸控制在4~20 nm。采用相同的方法可制备出各种金属-氧化铁哑铃型MNPs,包括Ag-Fe3O4[79]、AuAg-Fe3O4[79]、Pt-Fe3O4[80]、FePt-Fe3O4[81]和PtPd-Fe3O4 MNPs[82]。
哑铃型MNPs也可通过贵金属MNPs在预先形成的MNPs上成核和生长来制备。例如,在磁性铁氧体MFe2O4 MNPs存在下,通过油胺对乙酸银或硝酸银的还原可得哑铃型Ag-MFe2O4 MNPs[83]。可通过控制反应时间来对Ag NPs的尺寸进行控制。图 4(b)和图 4(c)是用这种方法制备得到的MNPs的TEM图。不同于结晶的MFe2O4 NPs种子,当无定形核/壳Fe/FexOy MNPs用作种子时,Ag在每个Fe/FexOy NP周围进行多位点成核和生长[84]。随着反应的进行,内部的Fe被进一步氧化,FexOy表面上的小Ag MNPs进一步成熟为更大的MNPs,产生具有空心FexOy的哑铃型Ag-FexOy MNPs。通过使用预先形成的双组分哑铃型MNPs作为种子,可以合成出更复杂的哑铃型MNPs。如在Fe3O4-Au-Fe3O4哑铃型MNPs的合成中,在S存在下将两个Au-Fe3O4 MNPs的Au侧面融合[76]。没有S的话,预先形成的Au-Fe3O4 MNPs十分稳定,但是在S的存在下,Au-S强键的形成使Au粒子不稳定,促使两个Au-Fe3O4 MNPs之间的两个Au粒子凝聚,因此形成了Fe3O4-Au-Fe3O4 MNPs。
MNPs,尤其是超顺磁MNPs,它们的生物医学应用已经得到了广泛的研究。由于外部磁场中的超顺磁行为和高磁矩,这些在生物环境中稳定的MNPs可以快速响应外部磁场,在每个MNP周围产生次级场,并干扰质子核自旋弛豫。这种MNPs可以用作磁共振成像(MRI)中的灵敏磁性探针(造影剂)。此外,当暴露于交变磁场(AMF)中时,它们可以快速切换其磁化方向,将磁能转化为热能,可以用于磁流体热疗(MFH)消除热敏癌细胞。
为了满足生物医学应用的要求,MNPs的表面需要是亲水性和生物相容性的。用于MNPs表面修饰的最常用方法有配体交换、配体加成、蛋白包裹和亲水性二氧化硅(SiO2)涂覆。
配体交换是用亲水配体直接取代MNPs上的疏水表面活性剂。亲水性配体通常由两个官能团组成,一个基团可牢固地结合到MNPs表面,另一个是亲水性的,使得MNPs可分散在水溶液中或进一步功能化。例如,对氧化铁MNPs的表面进行多巴胺修饰[85],多巴胺可以进一步用次氮基三乙酸进行功能化以进行蛋白质分离。将多巴胺与生物相容性聚合物配体(例如聚乙二醇(PEG))偶联提供了更强的空间排斥并改善了水溶液中的胶体稳定性。而且,MNPs上的PEG涂层可以减少它们与蛋白质的相互作用,使其对先天免疫系统“隐身”[86]。Nathan等[87]合成了三氟乙基氧羰基-聚(乙二醇)硅烷并在氧化铁MNPs上进行自组装。由此制备的MNPs具有通过羧基或胺基与细胞靶向剂共轭的灵活性,可用于许多生物医学应用,包括磁共振成像和受控药物递送。图 5(a)是该反应的示意图。
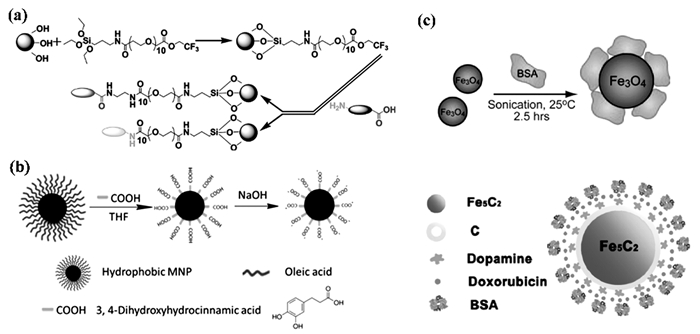
配体加成是另一种修饰策略。通过用两亲性配体包封这些MNPs可以将疏水性MNPs转移到亲水性介质中。两亲性配体的亲水性区域使得MNPs可以在水溶液中分散。通过将合成出的MNPs与两亲配体在适当的溶剂(例如氯仿)中混合,可以实现有效的配体添加[88]。Li等在单羧基封端的聚乙二醇(MPEG-COOH)存在下,在2-吡咯烷酮中热分解三乙酰基丙酮酸铁制备得到了生物相容性磁铁矿纳米晶。结果显示,MPEG通过COOH基团共价结合到纳米晶表面。MRI实验表明,由于其优异的水溶性和生物相容性,这些包被的纳米晶可以用作MRI造影剂。Liu等[89]开发了一种简单、高效、单相和低成本的方法,使用四氢呋喃、NaOH和3, 4-二羟基氢化肉桂酸将疏水性MNPs转化为亲水性MNPs,而没有任何复杂的有机合成(图 5(b))。所制备的亲水性MNPs在很宽的pH范围(pH=3~12)内是水溶性的,且溶解度是pH可控的。此外,带有羧酸根的MNPs可以容易地进一步表面官能化。该策略可以扩展到其他类型的疏水MNPs,以促进纳米材料的生物医学应用。
天然高分子,例如蛋白质,含有丰富的功能基团,生物相容性较好,适合用于MNPs的表面修饰。其中白蛋白是血浆中含量最多的蛋白质,具有无毒性及免疫原性、简单便宜、存储稳定性强等优点。既可直接与裸MNPs超声吸附,也可在蛋白包裹MNPs之前先用多巴胺等转水修饰(图 5(c))[90]。
SiO2涂层被广泛用于MNPs修饰。在SiO2涂覆之前,需要将疏水性MNPs从有机相转移至水相,通常使用亲水配体如十六烷基三甲基溴化铵(CTAB)[91]作为相转移剂,CTAB作为有机模板在每个MNP核周围形成介孔SiO2壳。疏水性Fe3O4 MNPs也可以在反相微乳液中被SiO2直接涂覆[92]。
MRI成像技术可以测量实体组织周围水分子与周围生物溶液的质子核磁共振差异。成像过程产生两种同时且独立的驰豫过程:纵向驰豫过程(T1)和横向驰豫过程(T2)。造影剂的效率通常由其弛豫速率ri(i=1, 2)表示,该值由引入造影剂后的弛豫速率变化量Δ(1/Ti)定义,单位是L·mmol-1·s-1,ri越大,MRI灵敏度越好。
促进T1弛豫的造影剂被称为T1造影剂。Gd3+顺磁性配合物是常见的T1造影剂,然而Gd配合物在生物系统中通常是短暂存在的,且游离的Gd3+可以从Gd配合物中浸出,引起不希望的毒性。Shen等[93]研发出了单分散MnO MNPs作为新的T1造影剂,然而这些MnO MNPs的r1值只有0.4 (mmol/L)-1·s-1,比基于Gd的T1造影剂(~4 (mmol/L)-1·s-1)还要低。Kim等[94]对超顺磁氧化铁MNPs作为T1造影剂进行了探索,他们研究了一系列尺寸为1.5、2.2、3和12 nm的氧化铁MNPs,该实验表明3nm氧化铁MNPs是优异的T1造影剂。Tromsdorf等[95]制备出了PEG包被的4nm氧化铁MNPs,该MNPs作为T1造影剂时r1高达7.3(mmol/L)-1·s-1,比标准的基于Gd的T1造影剂Magnevist高约两倍。Lu等[4]制备了聚乙二醇稳定的氧化铁纳米团簇(PEG-IONCs),研究了其作为T1 MRI造影剂在大型动物模型上的MRI成像应用。实验表明,PEG-IONCs具有高度的生物相容性,这些生物相容性PEG-IONCs成功地用于几种大型动物模型(包括兔子、狗和猕猴,图 6(a)和(b))中的MRI造影增强。
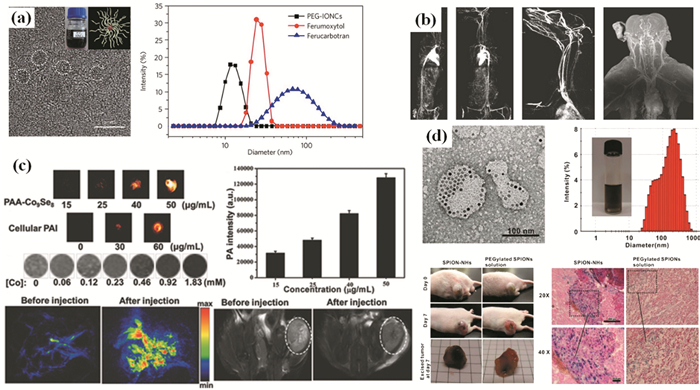
具有高Ms的超顺磁MNPs可显著缩短附近质子的T2弛豫时间并使T2加权MR图像变暗,这些超顺磁MNPs被称为T2造影剂。由于T2对比度效应强烈依赖于MNPs的磁化,可以增强超顺磁性MNPs的Ms来改善。单分散Fe3O4 MNPs作为研究MNPs尺寸效应对T2对比度的影响是一个很好的模型系统。对12nm MFe2O4(M=Ni,Co,Fe和Mn)MNPs的研究表明,具有较高Ms的MNPs表现出更好的T2对比度[12]。这些铁氧体MNPs(M=Ni,Co,Fe,Mn)的饱和磁化值分别为85、101、110 emu·g(Fe+M)-1,对应的r2分别为152、172、218和358 (mmol/L)-1·s-1。r2值越高,在T2加权MRI图像中MFe2O4 MNPs就显示出更高的对比效果。Zhou等[96]的研究指出,MNPs探针聚集体中局域磁场的不均匀性是影响T2弛豫效能的关键因素。氧化铁团簇和Landau-Lifshitz-Gilbert模拟的r2值证实了他们的假设,表明解决磁场的不均匀性可能成为建立MNPs(特别是磁性团簇)的磁化强度和T2弛豫率之间相关性的有效方法。他们首次利用磁场不均匀性因素阐明了单个探针和它们聚集体的MRI造影剂之间的相互关系,这将成为弥补探针聚集体的造影剂理论的空白,并为发展新型高效的MRI造影剂提供重要参考。
为了提高MRI的灵敏度和准确性,还可采用多功能MNPs的多模式成像探针。例如,Park等[97]制备出了涂覆有均匀SiO2的15nm MnFe2O4 MNPs作为T1-T2双模式造影剂,其上沉积了1.5nm厚的Gd2O(C4)2层,其中MnFe2O4MNPs用作T2造影剂,Gd2O(C4)2用作T1造影剂。将光学成像模式与MRI相结合是另一种改进方法。Song等[5]制备出了生物相容性的聚丙烯酸(PAA)-Co9Se8纳米片用于光声成像(PAI)/磁共振成像(MRI)双模式成像。在用PAA-Co9Se8纳米片处理的癌细胞中观察到比不含PAA-Co9Se8纳米片的癌细胞更亮的PAI图像,并且随着PAA-Co9Se8纳米片浓度增加获得的纳米片水溶液MRI图像较暗(图 6(c))。将PAA-Co9Se8纳米片溶液注射到携带HepG2肿瘤的小鼠,获得的PA图像在肿瘤区域周围显示出强烈的光声信号,用9.4T MR仪记录T2图像可观察到肿瘤部位明显的变黑效应。该结果证实,PAA-Co9Se8纳米片可能是用于PAI和MRI的有希望的双模态造影剂。
在AMF中,可以使用MNPs将磁能转换成热能。由于癌细胞比正常细胞对热更敏感,癌细胞周围的MNPs产生的热量可抑制癌细胞生长甚至消除这些癌细胞。这种癌症的物理治疗方法称之为磁流体热疗(MFH)。一般来说,MNPs可以通过以下三种磁性过程中的任何一种产生热量:磁滞损耗、Neel驰豫和布朗驰豫。对于单畴MNPs,热量由MNPs的Neel驰豫和布朗驰豫产生,加热效率由特定损耗功率(SLP)决定。在Rosensweig理论中,单分散MNPs的SLP随着AMF的强度和频率的增加而增加。然而,为了在电磁辐射下进行安全的临床热疗,需要仔细考虑AMF的应用强度和频率。
SLP强烈依赖于MNPs的Ms,Ms越高,SLP越高。因此可以使用高Ms的MNPs来增强SLP。Jang等[13]制备出了具有高Ms的Zn(Ⅱ)掺杂的铁氧体MNPs用于MFH。在体外试验中,当Zn0.4Mn0.6Fe2O4 MNPs用于治疗HeLa细胞时,发现10min的AMF暴露后消除了84.4%的癌细胞,而使用Feridex时,只有13.5%的癌细胞死亡。此外,Bae等[98]制备出了高生物相容性的镁掺杂的γ-Fe2O3(Mg0.13-γ-Fe2O3)MNPs,该MNPs具有特别高的固有损耗功率(14 nH·m2·kg-1),比商业Fe3O4(Feridex,0.15 nH·m2·kg-1)高出近100倍,他们发现Mg0.13-γ-Fe2O3的热诱导特性明显增强,具有很好的高温效应,能够完全杀死肿瘤细胞。
磁各向异性在SLP增强中也起着关键作用。以铁氧体MNPs为例,K可以很容易地通过不同的过渡金属掺杂来调节,如CoxFe3-xO4系统,单分散CoFe2O4 MNPs展现出比Fe3O4 MNPs(152W·g-1)高3倍的的SLP(443W·g-1)[99]。优化SLP应该考虑同时考虑K和Ms。一个典型的例子是由硬磁的CoFe2O4核和软磁的MnFe2O4外壳组成的交换耦合核/壳MNPs[99]。15nm CoFe2O4/MnFe2O4 MNPs在室温下保持了超顺磁性,K值为1.5×104J·m-3,处于最佳K值范围。与单组分MNPs(CoFe2O4 MNPs为443W·g-1,MnFe2O4 MNPs为411W·g-1)相比,核/壳MNPs的SLP值为2280W·g-1,高出5倍。
与单次MFH诱导细胞凋亡相比,使用多重MFH也可通过坏死途径有效杀死癌细胞。Zhang等[6]制备了可注射的、生物可降解、热敏和载有超顺磁氧化铁纳米粒子的纳米胶囊水凝胶(SPION-NHs)(图 6(d)),并将其用于多重MFH和长期的磁共振成像。通过T2加权MRI监测到,单次注射SPION-NHs后,肿瘤内SPIONs的保留时间超过3周,极大促进了多重MFH疗效。与单次MFH相比,四重MFH治疗显示了优异的活体抗肿瘤效果,也避免了周围健康组织的严重损伤。
本文总结了MNPs的高温有机相合成方法及其在生物医学中的应用进展。主要关注的是尺寸在60nm以内的MNPs,尺寸依赖的磁性能可以对其进行调整和优化,从而适用于多种重要的技术应用。
近年来,高温有机相合成已被广泛研究用于制备MNPs。有机相的高沸点允许反应在高温下进行,从而促进具有更高程度晶体有序性的元素金属、合金或更复杂的MNPs的成核和生长。化学惰性有机溶剂对于MNPs表面官能化和稳定作用的表面活性剂也是有益的。通过调整表面活性剂的化学性质,可以控制MNPs的尺寸和形状。有机相合成特别适用于制备复合MNPs,如核/壳和哑铃型MNPs,从而可以将不同的功能引入到MNPs中。目前,科学家正在积极将MNPs的有机相合成扩大到工业规模。考虑到不同实验室使用的化学品质量差异很大,可能会导致重复合成的困难,现阶段最紧迫的问题可能是统一和改善这些化学品的质量。一旦解决这个问题,这些单分散MNPs的配方应该很容易被重复和放大以便大量生产。
由于具有强的磁响应以及生物环境中的稳定性,超顺磁性MNPs有着重要的生物医学应用。由有机相合成制备的MNPs可以通过配体交换、表面活性剂添加或SiO2涂覆进行表面修饰,形成稳定的分散体。表面修饰不仅使这些MNPs在生物系统中稳定,还能促进它们与生物靶向剂的进一步功能化。超顺磁MNPs可以作为MRI的灵敏探针,复合的MNPs作为多模式成像的探针更为重要,可以同时进行MRI和其他模态成像,提高灵敏度和准确性。MNPs还可以作为MFH的高效热疗剂,具有可控的磁加热效率。目前的研究重点是研究功能化MNPs与生物分子及细胞之间的相互作用,以便更好地了解这些MNPs在生物系统中的生物循环、生物分布和生物去除,并研究靶向特异性用于灵敏诊断和治疗。总之,有机相合成单分散MNPs的最新进展不仅有助于深入了解其磁学特性,而且也为重要纳米生物技术应用的未来设计和先进制造铺平了道路。
L Wu, A Mendoza-Garcia, Q Li et al. Chem. Rev., 2016, 116: 10473~10512. doi: 10.1021/acs.chemrev.5b00687
D Ho, X Sun, S Sun. Acc. Chem. Res., 2011, 44: 875~882. doi: 10.1021/ar200090c
L H Reddy, J L Arias, J Nicolas et al. Chem. Rev., 2012, 112: 5818~5878. doi: 10.1021/cr300068p
Y Lu, Y J Xu, G B Zhang et al. Nat.Biomed. Eng., 2017, 1: 637~643. doi: 10.1038/s41551-017-0116-7
X R Song, X Wang, S X Yu et al. Adv. Mater., 2015, 27: 3285~3291. doi: 10.1002/adma.201405634
Z Q Zhang, S C Song. Biomaterials, 2016, 106: 13~23. doi: 10.1016/j.biomaterials.2016.08.015
R Skomski, J M D Coey. Permanent Magnetism, 1999.
L Néel. C. R. Acad. Sci., 1949, 228: 664~666.
C M Sorensen. Magnetism, in Nanoscale Materials in Chemistry, 2001.
Y Soumare, C Garcia, T Maurer et al. Adv. Funct. Mater., 2009, 19: 1971~1977. doi: 10.1002/adfm.200800822
G Cherubini, R D Cideciyan, L Dellmann et al. IEEE Trans. Magn., 2011, 47: 137~147. doi: 10.1109/TMAG.2010.2076797
J H Lee, Y M Huh, Y Jun et al. Nat. Med., 2007, 13: 95~99. doi: 10.1038/nm1467
J T Jang, H Nah, J H Lee et al. Angew. Chem. Int. Ed., 2009, 48: 1234~1238. doi: 10.1002/anie.200805149
V K LaMer, R H Dinegar. J. Am. Chem. Soc., 1950, 72: 4847~4854. doi: 10.1021/ja01167a001
J Park, J Joo, S G Kwon et al. Angew. Chem. Int. Ed., 2007, 46: 4630~4660. doi: 10.1002/anie.200603148
S Peng, C Wang, J Xie et al. J. Am. Chem. Soc., 2006, 128: 10676~10677. doi: 10.1021/ja063969h
C T Black, C B Murray, R L Sandstrom et al. Science, 2000, 290: 1131~1134. doi: 10.1126/science.290.5494.1131
J Park, E Kang, S U Son et al. Adv. Mater., 2005, 17: 429~434. doi: 10.1002/adma.200400611
S H Sun, H Zeng. J. Am. Chem. Soc., 2002, 124: 8204~8205. doi: 10.1021/ja026501x
D Farrell, S A Majetich, J P Wilcoxon. J. Phys. Chem. B, 2003, 107: 11022~11030. doi: 10.1021/jp0351831
L M Lacroix, N F Huls, D Ho et al. Nano Lett., 2011, 11: 1641~1645. doi: 10.1021/nl200110t
A Meffre, B Mehdaoui, V Kelsen et al. Nano Lett., 2012, 12: 4722~4728. doi: 10.1021/nl302160d
F Dumestre, B Chaudret, C Amiens et al. Science, 2004, 303: 821~823. doi: 10.1126/science.1092641
L M Lacroix, S Lachaize, A Falqui et al. J. Am. Chem. Soc., 2009, 131: 549~557. doi: 10.1021/ja805719c
C B Murray, S H Sun, W Gaschler et al. IBM J. Res. Dev., 2001, 45: 47~56. doi: 10.1147/rd.451.0047
S Peng, J Xie, S H Sun. J. Solid State Chem., 2008, 181: 1560~1564. doi: 10.1016/j.jssc.2008.03.024
C Petit, A Taleb, M P Pileni. J. Phys. Chem. B, 1999, 103: 1805~1810. doi: 10.1021/jp982755m
Z J Yang, M Cavalier, M Walls et al. J. Phys. Chem. C, 2012, 116: 15723~15730. doi: 10.1021/jp303182n
D P Dinega, M G Bawendi. Angew. Chem. Int. Ed., 1999, 38: 1788~1791. doi: 10.1002/(SICI)1521-3773(19990614)38: 12 < 1788: : AID-ANIE1788 > 3.0.CO; 2-2
S H Sun, C B Murray. J. Appl. Phys., 1999, 85: 4325~4330. doi: 10.1063/1.370357
O Metin, V Mazumder, S Ozkar et al. J. Am. Chem. Soc., 2010, 132: 1468~1469. doi: 10.1021/ja909243z
M Cargnello, V V T Doan-Nguyen, T R Gordon et al. Science, 2013, 341: 771~773. doi: 10.1126/science.1240148
N Cordente, M Respaud, F Senocq et al. Nano Lett., 2001, 1: 565~568. doi: 10.1021/nl0100522
A P LaGrow, B Ingham, S Cheong et al. J. Am. Chem. Soc., 2012, 134: 855~858. doi: 10.1021/ja210209r
A Hutten, D Sudfeld, I Ennen et al. J. Magn. Magn. Mater., 2005, 293: 93~101. doi: 10.1016/j.jmmm.2005.01.048
C Desvaux, C Amiens, P Fejes et al. Nat. Mater., 2005, 4: 750~753. doi: 10.1038/nmat1480
G S Chaubey, C Barcena, N Poudyal et al. J. Am. Chem. Soc., 2007, 129: 7214~7215. doi: 10.1021/ja0708969
C Wang, S Peng, L M Lacroix et al. Nano Res., 2009, 2: 380~385. doi: 10.1007/s12274-009-9037-4
S H Sun, C B Murray, D Weller et al. Science, 2000, 287: 1989~1992. doi: 10.1126/science.287.5460.1989
M Chen, J P Liu, S H Sun. J. Am. Chem. Soc., 2004, 126: 8394~8395. doi: 10.1021/ja047648m
L E M Howard, H L Nguyen, S R Giblin et al. J. Am. Chem. Soc., 2005, 127: 10140~10141. doi: 10.1021/ja051669e
S H Sun, S Anders, T Thomson et al. J. Phys. Chem. B, 2003, 107: 5419~5425.
E V Shevchenko, D V Talapin, A L Rogach et al. J. Am. Chem. Soc., 2002, 124: 11480~11485. doi: 10.1021/ja025976l
J I Park, J Cheon. J. Am. Chem. Soc., 2001, 123: 5743~5746. doi: 10.1021/ja0156340
W R Lee, M G Kim, J R Choi et al. J. Am. Chem. Soc., 2005, 127: 16090~16097. doi: 10.1021/ja053659j
M Chen, J Kim, J P Liu et al. J. Am. Chem. Soc., 2006, 128: 7132~7133. doi: 10.1021/ja061704x
Y L Hou, H Kondoh, T Kogure et al. Chem. Mater., 2004, 16: 5149~5152. doi: 10.1021/cm048902c
Y S Yu, K W Sun, Y Tian et al. Nano Lett., 2013, 13: 4975~4979. doi: 10.1021/nl403043d
G Jiang, H Zhu, X Zhang et al. ACS Nano, 2015, 9: 11014~11022. doi: 10.1021/acsnano.5b04361
K Ono, Y Kakefuda, R Okuda et al. J. Appl. Phys., 2002, 91: 8480~8482. doi: 10.1063/1.1456407
H W Gu, B Xu, J C Rao et al. J. Appl. Phys., 2003, 93: 7589~7591. doi: 10.1063/1.1537697
Y L Hou, Z C Xu, S Peng et al. Adv. Mater., 2007, 19: 3349~3352. doi: 10.1002/adma.200700891
G S Chaubey, N Poudyal, Y Z Liu et al. J. Alloys Compd., 2011, 509: 2132~2136. doi: 10.1016/j.jallcom.2010.10.164
S H Sun, H Zeng, D B Robinson et al. J. Am. Chem. Soc., 2004, 126: 273~279. doi: 10.1021/ja0380852
H Zeng, P M Rice, S X Wang et al. J. Am. Chem. Soc., 2004, 126: 11458~11459. doi: 10.1021/ja045911d
Z C Xu, C M Shen, Y L Hou et al. Chem. Mater., 2009, 21: 1778~1780. doi: 10.1021/cm802978z
C Moya, O Iglesias-Freire, N Perez et al. Nanoscale, 2015, 7: 8110~8114. doi: 10.1039/C5NR00592B
T Hyeon, S S Lee, J Park et al. J. Am. Chem. Soc., 2001, 123: 12798~12801. doi: 10.1021/ja016812s
N Z Bao, L M Shen, Y H Wang et al. J. Am. Chem. Soc., 2007, 129: 12374~12375. doi: 10.1021/ja074458d
Z Zhao, Z Zhou, J Bao et al. Nat Commun., 2013, 4: 2266. doi: 10.1038/ncomms3266
I S Lee, N Lee, J Park et al. J. Am. Chem. Soc., 2006, 128: 10658~10659. doi: 10.1021/ja063177n
Y D Yin, R M Rioux, C K Erdonmez et al. Science, 2004, 304: 711~717. doi: 10.1126/science.1096566
H Zeng, J Li, Z L Wang et al. Nano Lett., 2004, 4: 187~190. doi: 10.1021/nl035004r
H Zeng, S H Sun, J Li et al. Appl. Phys. Lett., 2004, 85: 792~794. doi: 10.1063/1.1776632
F Liu, J H Zhu, W L Yang et al. Angew. Chem. Int. Ed., 2014, 53: 2176~2180. doi: 10.1002/anie.201309723
Q Song, Z J Zhang. J. Am. Chem. Soc., 2012, 134: 10182~10190. doi: 10.1021/ja302856z
S H Noh, W Na, J T Jang et al. Nano Lett., 2012, 12: 3716~3721. doi: 10.1021/nl301499u
T J Yoon, H Lee, H L Shao et al. Angew. Chem. Int. Ed., 2011, 50: 4663~4666. doi: 10.1002/anie.201100101
T J Zhou, M H Lu, Z H Zhang et al. Adv. Mater., 2010, 22: 403~406. doi: 10.1002/adma.200901801
N H Cho, T C Cheong, J H Min et al. Nat. Nanotechnol., 2011, 6: 675~682. doi: 10.1038/nnano.2011.149
H Kim, M Achermann, L P Balet et al. J. Am. Chem. Soc., 2005, 127: 544~546. doi: 10.1021/ja047107x
J S Lee, M I Bodnarchuk, E V Shevchenko et al. J. Am. Chem. Soc., 2010, 132: 6382~6391. doi: 10.1021/ja100029s
Q W Tian, J Q Hu, Y H Zhu et al. J. Am. Chem. Soc., 2013, 135: 8571~8577. doi: 10.1021/ja4013497
E V Shevchenko, M I Bodnarchuk, M V Kovalenko et al. Adv. Mater., 2008, 20: 4323~4329. doi: 10.1002/adma.200702994
J H Gao, G L Liang, B Zhang et al. J. Am. Chem. Soc., 2007, 129: 1428~1433. doi: 10.1021/ja067785e
W L Shi, H Zeng, Y Sahoo et al. Nano Lett., 2006, 6: 875~881. doi: 10.1021/nl0600833
W I Liang, X Zhang, Y Zan et al. J. Am. Chem. Soc., 2015, 137: 14850~14853. doi: 10.1021/jacs.5b10076
H Yu, M Chen, P M Rice et al. Nano Lett., 2005, 5: 379~382. doi: 10.1021/nl047955q
C Wang, H F Yin, S Dai et al. Chem. Mater., 2010, 22: 3277~3282. doi: 10.1021/cm100603r
C Wang, H Daimon, S H Sun. Nano Lett., 2009, 9: 1493~1496. doi: 10.1021/nl8034724
Q Li, L Wu, G Wu et al. Nano Lett., 2015, 15: 2468~2473. doi: 10.1021/acs.nanolett.5b00320
X L Sun, S J Guo, C S Chung et al. Adv. Mater., 2013, 25: 132~136. doi: 10.1002/adma.201203218
L Zhang, Y H Dou, H C Gu. J. Colloid Interf. Sci., 2006, 297: 660~664. doi: 10.1016/j.jcis.2005.11.009
S Peng, C H Lei, Y Ren et al. Angew. Chem. Int. Ed., 2011, 50: 3158~3163. doi: 10.1002/anie.201007794
C J Xu, K M Xu, H W Gu et al. J. Am. Chem. Soc., 2004, 126: 9938~9939. doi: 10.1021/ja0464802
A S Karakoti, S Das, S Thevuthasan et al. Angew. Chem. Int. Ed., 2011, 50: 1980~1994. doi: 10.1002/anie.201002969
N Kohler, G E Fryxell, M Zhang. J. Am. Chem. Soc., 2004, 126: 7206~7211. doi: 10.1021/ja049195r
J M Shin, R M Anisur, M K Ko et al. Angew. Chem. Int. Ed., 2009, 48: 321~324. doi: 10.1002/anie.200802323
Y Liu, T Chen, C C Wu et al. J. Am. Chem. Soc., 2014, 136: 12552~12555. doi: 10.1021/ja5060324
C Qian, L Zhuang. Adv. Mater., 2016, 28: 10557~10566. doi: 10.1002/adma.201600038
J Kim, H S Kim, N Lee et al. Angew. Chem. Int. Ed., 2008, 47: 8438~8441. doi: 10.1002/anie.200802469
A Guerrero-Martinez, J Perez-Juste, L M Liz-Marzan. Adv. Mater., 2010, 22: 1182~1195. doi: 10.1002/adma.200901263
H B Na, J H Lee, K J An et al. Angew. Chem. Int. Ed., 2007, 46: 5397~5401. doi: 10.1002/anie.200604775
B H Kim, N Lee, H Kim et al. J. Am. Chem. Soc., 2011, 133: 12624~12631. doi: 10.1021/ja203340u
U I Tromsdorf, O T Bruns, S C Salmen et al. Nano Lett., 2009, 9: 4434~4440. doi: 10.1021/nl902715v
Z Zhou, R Tian, Z Wang et al. Nat Commun., 2017, 8: 15468. doi: 10.1038/ncomms15468
J S Choi, J H Lee, T H Shin et al. J. Am. Chem. Soc., 2010, 132: 11015~11017. doi: 10.1021/ja104503g
J T Jang, J Lee, J Seon et al. Adv. Mater., 2018, 30: 1704362. doi: 10.1002/adma.201704362
J H Lee, J T Jang, J S Choi et al. Nat. Nanotechnol., 2011, 6: 418~422. doi: 10.1038/nnano.2011.95



图 3 (a) 四种不同Fe3-xO4 MNPs的AFM图(上)和MFM图(下)[57];(b)不同边缘长度的八足体氧化铁MNPs及其XPS谱图和模型[60]
Figure 3 (a) Topographic AFM (top row) and the corresponding MFM images (bottom row) of four different Fe3-xO4 NPs (Reprinted from ref 57); (b) The octapod iron oxide nanoparticles with edge lengths and its X-ray photoelectron spectroscopy analysis and its model illustration[60]


图 5 (a) 三氟乙基氧羰基-末端聚(乙二醇)(PEG)硅烷在MNPs上的固定[87];(b)使用3, 4-二羟基氢化肉桂酸(DHAC)、NaOH、THF进行配体交换[89];(c)使用白蛋白表面修饰的两种MNPs[90]
Figure 5 (a) Immobilization of PEG on magnetite nanoparticles; (b) Ligand exchange using 3, 4-dihydroxyhydrocinnamic acid (DHAC), NaOH and tetrahydrofuran; (c) Scheme showing the use of albumin for surface modification of two types of MNPs

图 6 (a) PEG-IONCs的表征;TEM图(左)和动态光散射数据(右)[4];(b)使用PEG-IONCs的比格犬和猕猴的MRI图像,显示出脉管系空间分辨率[4];(c)PAA-Co9Se8纳米片表征以及在用PAA-Co9Se8纳米片注射前后的肿瘤部位的PA图像和代表性的鼠的T2加权MRI扫描[5];(d)SPION-NHs的表征及其在体内的保留行为[6]
Figure 6 (a) Characterization of the PEG-IONCs. High-resolution TEM image (left) and the Dynamic light scattering data(right)[4]; (b) MRI of a beagle dog and a macaque using PEG-IONCs, showing spatial resolution of the vasculature[4]; (c) characterization of PAA-Co9Se8 nanoplates and the PA images of tumor site before and after injection with PAA-Co9Se8 nanoplates and the representative T2-weighted MRI scans of mice[5]; (d) Characterization of SPION-NHs and the in vivo retention behaviors of SPION-NHs[6]
表 1 MNPs的合成方法及其性能
Table 1. Synthesis methods and properties of MNPs
| 成分 | 反应试剂 | 表面活性剂 | 溶剂 | 形貌尺寸 | 磁性能 | 参考文献 |
| Fe | Fe(CO)5 | 油胺 | 1-十八烯 | 球形, < 10nm | Ms=102.6emu·g-1 | [16] |
| Fe(CO)5 | 油酸,油胺 | 二辛基醚 | 球形,5~20nm | Ms=173emu·g-1 | [20] | |
| Fe(CO)5 | 油胺,十六烷基氯化铵 | 1-十八烯 | 立方形,15nm | Ms=180emu·g-1, Hc=800Oe (5K) |
[21] | |
| Fe[N(SiMe3)2]2 | 油酸,十六烷基胺 | 均三甲苯 | 立方形,~6.2nm | Mr=83 emu·g-1 Ms=212 emu·g-1 |
[23] | |
| Fe[N(SiMe3)2]2 | 棕榈酸,十六烷基胺 | 均三甲苯 | 方形,13~20nm | - | 24 | |
| Co | Co2(CO)8 | 油酸,二正辛胺 | 二苯醚 | 核壳结构,球形,8~14nm | Ms=125 emu·g-1 | [26] |
| CoCl2(超氢化物) | 油酸,三烷基膦 | 二辛基醚 | 球形纳米晶,2~11nm | Hc=500 Oe (5K) | [30] | |
| Ni | NiAc2 | 油酸,三丁基膦 | 二苯醚 | 球形, < 6nm | Hc=3000~5000 Oe | [31] |
| Ni(acac)2 | 油胺,三正辛基膦 | 苄基醚 | 六角超晶格,4~12nm | - | [32] | |
| Ni(COD)2 | 十六烷基胺, 三正辛基氧膦 | 四氢呋喃 | 纳米棒,4×15nm | Ms=60 emu·g-1, Hc=275 Oe (2K) |
[33] | |
| Ni(acac)2 | 三正辛膦, 十六烷基胺 | 均三甲基苯 | 纳米立方体,12nm | Hc=105 Oe (10K), Mr=3.2 emu·g-1, Ms=12.4 emu·g-1 |
[34] | |
| FeCo | Fe(CO)5, Co2(CO)8 | 油酸,三辛基膦 | 1, 2-二氯苯 | 球形,1~11nm | Hc=17 Oe (室温) | [35] |
| Fe(CO)5 Co(N(SiMe3)2)2 | 油酸,十六烷基胺 | 甲苯 | 球形,15nm | Ms=160A·m2·kg-1 | [36] | |
| Fe(acac)3, Co(acac)2 | 油酸,油胺 | 油胺 | 球形,20nm | Ms=207 emu·g-1 Hc=300 Oe (室温) |
[37] | |
| FePt | Fe(CO)5,Pt(acac)2 | 油酸,油胺 | 二辛基醚 | 六角密堆积超晶格,3~10nm | Hc=180 Oe(5K) | [39] |
| Fe(CO)5, Pt(acac)2 | 油酸,油胺 | 苯醚 | 纳米立方体,3~9nm | Hc=22 kOe (室温) | [46] | |
| Na2Fe(CO)4, Pt(acac)2 | 油酸 | 十九烷 | 面心立方晶体,<3nm, | Hc=3100 Oe (10K) | [41] | |
| FeCl2, Pt(acac)2/ superhydride |
油酸,油胺 | 二苯醚 | 球形,4nm | Hc=2400 Oe (室温) | [42] | |
| CoPt3 | Co2(CO)8,Pt(acac)2 | 1-金刚烷甲酸,十六胺 | 二苯醚 | 球形纳米晶,1.5 ~7.2nm | - | [43] |
| Co2(CO)8,Pt(hfac)2 | 油酸 | 甲苯 | 球形,<10nm | Hc=6900 Oe(5K) | [44] | |
| Co2(CO)8,Pt(hfac)2 | 十二烷异腈 | 壬烷 | 球形核壳结构,4.7nm | Hc=660 Oe(5K) | [45] | |
| FePd | Fe(CO)5,Pd(acac)2 | 1-金刚烷羧酸,三烷基膦 | 油酸 | 球形,11~16nm | Hc=350 Oe(2K) | [47] |
| FePd-Fe3O4 | Fe(CO)5,Pd(acac)2 | 油酸,油胺 | 1-十八烯 | 海胆状,60nm | Hc=0.8~2.6 kOe, Ms=90~190 emu·g-1 |
[48] |
| FePd/Pd | Fe(CO)5,Pd(acac)2 | 油胺 | 四氢化萘 | 核壳结构, 面心四方, 8.5nm | Ms=169 emu·g-1, Hc=1.0 kOe(室温) |
[49] |
| SmCo5 | Co2(CO)8,Sm(acac)3 | 油酸 | 二辛基醚 | 面心立方晶体,9nm | Hc=50Oe(300K) | [50] |
| Co2(CO)8,Sm(acac)3 | 油酸,油胺, 十六烷二醇 | 二辛基醚 | 球形,6~8nm | Hc=2.2 kOe(5K) | [51] | |
| Co2(CO)8,Sm(acac)3 | 油酸,二正辛胺 | 四氢化萘 | 球形,32nm | Hc=8 kOe(室温), Mr=40~50 emu·g-1 |
[52] | |
| Co(acac)2,Sm(acac)3 | 油酸, 三辛膦, 十六烷二醇 | 油酸 | 球形,25nm | Hc=12 kOe(室温), Ms=70 emu·g-1 |
[53] | |
| Fe3O4 | Fe(acac)3 | 油胺 | 苯醚 | 球形,7~10nm | Ms=80 emu·g-1 | [19] |
| Fe(acac)3 | 油胺,油酸 | 二苄醚 | 球形,3~20nm | Ms=82 emu·g-1 | [54] | |
| Fe3-xO4 | Fe(acac)3 | 癸酸 | 二苄醚 | 立方体,30nm | Ms=80~83 emu·g-1 | [57] |
| FePt-Fe3O4 | Pt(acac)2, Fe(CO)5, Fe(acac)3 | 油酸,油胺 | 辛基醚、二苯醚 | 核壳结构, 核4nm, 壳0.5~3nm | Hc=5.5 kOe(10K) | [63] |
| MnFe2O4/Fe | Fe(CO)5, Fe(acac)3, Mn2(CO)10 | 油酸,油胺 | 1-十八烯 | 核壳结构, < 8nm | Ms=149emu·g-1 | [68] |
| Fe3O4/Au/Fe3O4 | Fe(CO)5, HAuCl4 | 油酸,油胺 | 1-十八烯 | 哑铃型,Au 8nm, Fe3O4 20nm | Ms=80 emu·g-1 | [76] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们