引用本文:
陈佳敏, 周长松, 杨宏旻, 吴昊. Mo/Fe3O4(111)表面对燃煤烟气汞吸附的密度泛函研究[J]. 燃料化学学报,
2020, 48(5): 525-532.
Citation:
CHEN Jia-min, ZHOU Chang-song, YANG Hong-min, WU Hao. A DFT study on the adsorption of various mercury species in the coal combustion flue gases on the Mo-doped Fe3O4(111) surface[J]. Journal of Fuel Chemistry and Technology,
2020, 48(5): 525-532.
Received Date:
19 January 2020 Revised Date:
16 March 2020 Available Online:
01 May 2020
Fund Project:
The project was supported by the National Natural Science Foundation of China (51676101, 51806107) and the Natural Science Foundation of Jiangsu Province(BK20161558, BK20180731)
Abstract:
The adsorption characteristics of Hg0, HgCl and HgCl2 on the Mo-doped Fe3O4 (111) Fetet surface were investigated by density functional theory (DFT) calculation with the CASTEP software package. The results indicate that both HgCl and HgCl2 are chemically adsorbed on the Mo-doped Fe3O4 (111) Fetet surface, whereas Hg0 is bound to the surface by physisorption. The binding energies of HgCl on the Mo-doped Fe3O4 (111) Fetet surface is about 40%-66% higher than that on the pure Fe3O4 (111) Fetet surface. For the adsorption of HgCl2 molecule on the pure Fe3O4 (111) Fetet surface, two Cl atoms interact with one Mo atom and one Fe atom, forming the "M" structure; in contrast, on the Mo-doped Fe3O4 (111) Fetet surface, the stronger interaction between Cl atom and Mo atom allows a complete dissociation of HgCl2 and release of Hg. The adsorption mechanism of mercury species on the Mo-doped Fe3O4 (111) Fetet surface revealed in this work may be helpful for the practical removal of mercury from coal-fired flue gases.
中国统计年鉴[M].北京: 中国统计出版社, 2009.Statistical Yearbook of China[M]. Beijing: China Statistics Press, 2009
[2]
ZHENG Y, JENSEN A D, WINDELIN C, JENSEN F. Review of technologies for mercury removal from flue gas from cement production processes[J]. Prog Energy Combust,
2012, 38(5):
599-629.
doi: 10.1016/j.pecs.2012.05.001
[3]
CARPI A. Mercury from combustion sources:A review of the chemical species emitted and their transport in the atmosphere[J]. Water Air Soil Poll,
1997, 98(3/4):
241-254.
doi: 10.1023/A:1026429911010
[4]
YANG Y, LIU J, WANG Z, ZHANG Z. Homogeneous and heterogeneous reaction mechanisms and kinetics of mercury oxidation in coal-fired flue gas with bromine addition[J]. Proc Combust Inst,
2016, 36(3):
4039-4049.
doi: 10.1016/j.proci.2016.08.068
[5]
ZHANG B K, LIU J, ZHENG C G, CHANG M. Theoretical study of mercury species adsorption mechanism on MnO2(110) surface[J]. Chem Eng J,
2014, 256:
93-100.
doi: 10.1016/j.cej.2014.07.008
[6]
GALBREATH K C, ZYGARLICKE C J. Mercury speciation in coal combustion andgasfication flue gases[J]. Environ Sci Technol,
1996, 30(8):
2421-2426.
doi: 10.1021/es950935t
[7]
WHO (World Health Organization). Preventing Disease through Healthy Environment: Exposure to Mercury: A Major Public Health Concern[R]. Geneva, Switzerland, WHO, 2007.https://www.who.int/ipcs/features/mercury.pdf
[8]
SJOSTROM S, DURHAM M, BUSTARD C J, MARTIN C. Activated carbon injection for mercury control:overview[J]. Fuel,
2010, 89(6):
1320-1322.
doi: 10.1016/j.fuel.2009.11.016
[9]
YANG H, XU Z, FAN M, BLAND A E, JUDKINS R R. Adsorbents for capturing mercury in coal-fired boiler flue gas[J]. J Hazard Mater,
2017, 146(1/2):
1-11.
[10]
WANG P, HU S, XIANG J, SU S, SUN L, CAO F, XIAO X, ZHANG A. Analysis of mercury species over CuO-MnO2-Fe2O3/γ-Al2O3 catalysts by thermal desorption[J]. Proc Combust Inst,
2015, 35(3):
2847-2853.
doi: 10.1016/j.proci.2014.06.054
[11]
ZHANG A, ZHENG W, SONG J, HU S, LIU Z, XIANG J. Cobalt manganese oxides modified titania catalysts for oxidation of elemental mercury at low flue gas temperature[J]. Chem Eng J,
2014, 236:
29-38.
doi: 10.1016/j.cej.2013.09.060
[12]
ZHOU C S, SUN L S, XIANG J, HU S, SU S, ZHAO A C. The experimental and mechanism study of novel heterogeneous Fenton-like reactions using Fe3-xTixO4 catalysts for Hg0 absorption[J]. Proc Combust Inst,
2015, 35(3):
2875-2882.
doi: 10.1016/j.proci.2014.06.049
[13]
ZHOU C S, SUN L S, ZHANG A C, WU X F, MA C, SU S, HU S, XIANG J. Fe3-xCuxO4 as highly active heterogeneous Fenton-like catalysts towards elemental mercury removal[J]. Chemosphere,
2015, 125:
16-24.
doi: 10.1016/j.chemosphere.2014.12.082
[14]
LI H L, FENG S H, YANG Z Q, YANG J P, LIU S J, HU Y C, ZHONG L, QU W Q. Density functional theory study of mercury adsorption on CuS surface:Effect of typical flue gas components[J]. Energy Fuels,
2019, 33(2):
1540-1546.
doi: 10.1021/acs.energyfuels.8b03585
[15]
ZHAO H T, MU X L, YANG G, GEORGE M. Graphene-like MoS2 containing adsorbents for Hg0 capture at coal-fired power plants[J]. Appl Energy,
2017, 207:
254-264.
doi: 10.1016/j.apenergy.2017.05.172
[16]
WANG Z, LIU J, YANG Y J, LIU F, DING J Y. Heterogeneous reaction mechanism of elemental mercury oxidation by oxygen species over MnO2 catalyst[J]. Proc Combust Inst,
2019, 37(3):
2967-2975.
doi: 10.1016/j.proci.2018.06.132
[17]
杨涛, 温晓东, 任君, 李永旺, 王建国, 霍春芳. Fe3O4(111), (110)和(001)表面结构的密度泛函理论研究[J]. 燃料化学学报,
2010,38,(1): 121-128.
doi: 10.3969/j.issn.0253-2409.2010.01.022YANG Tao, WEN Xiao-dong, REN Jun, LI Yong-wang, WANG Jian-guo, HUO Chun-fang. Surface structures of Fe304(111), (110), and (001):A density functional theory study[J]. J Fuel Chem Technol,
2010, 38(1):
121-128.
doi: 10.3969/j.issn.0253-2409.2010.01.022
[18]
YANG T, WEN X D, HUO C F, LI Y W, WANG J G, JIAO H J. Structure and energetics of hydrogen adsorption on Fe3O4(111)[J]. J Mol Catal A Chem,
2009, 302(1):
129-136.
[19]
YU X H, HUO C F, LI Y W, WANG J G, JIAO H J. Fe3O4 surface electronic structures and stability from GGA+U[J]. Surf Sci,
2012, 606(9/10):
872-879.
[20]
赵中霞, 任韧, 任奕璟, 周志立. 过渡元素掺杂Fe3O4(001)表面磁电性能的理论研究[J]. 无机化学学报,
2017,33,(6): 923-931.
ZHAO Zhong-xia, REN Ren, REN Yi-jing, ZHOU Zhi-li. Theoretical study of the magnetic and electric properties of transition elements doped Fe3O4(001) surface[J]. Chin J Inorg Chem,
2017, 33(6):
923-931.
[21]
FU Z M, YANG B W, ZHANG Y, ZHANG N, YANG Z X. Dopant segregation and CO adsorption on doped Fe3O4(111) surfaces:A first principle study[J]. J Catal,
2108, 364:
291-296.
doi: 10.1016/j.jcat.2018.05.027
[22]
FU Z M, WANG J Q, ZHANG N, AN Y P, YANG Z X. Effect of Cu doping on the catalytic activity of Fe3O4 in water-gas shift reactions[J]. Int J Hydrogen Energy,
2015, 40(5):
2193-2198.
doi: 10.1016/j.ijhydene.2014.12.063
[23]
XUE P Y, FU Z M, YANG Z X. The density functional theory studies on the promoting effect of the Cu-modified Fe3O4 catalysts[J]. Phys Lett A,
2015, 379(6):
607-612.
doi: 10.1016/j.physleta.2014.12.014
[24]
SONG Z J, WANG B, YU J, MA C, CHEN T, YANG W, LIU S, SUN L S. Effect of Ti doping on heterogeneous oxidation of NO over Fe3O4 (111) surface by H2O2:A density functional study[J]. Chem Eng J,
2018, 354:
517-524.
doi: 10.1016/j.cej.2018.08.042
PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phys Rev Lett,
1996, 77:
3865-3868.
doi: 10.1103/PhysRevLett.77.3865
[27]
WHITE J A, BIRD D M. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations[J]. Phys Rev B,
1994, 50(7):
4954-4957.
doi: 10.1103/PhysRevB.50.4954
[28]
周长松, 杨宏旻, 孙佳兴, 祁东旭, 毛琳, 宋子健, 孙路石. Fe3O4协同H2O2气相高级氧化单质汞的机理[J]. 化工学报,
2018,69,(5): 1840-1845.
ZHOU Chang-song, YANG Hong-min, SUN Jia-xing, QI Dong-xu, MAO Lin, SONG Zi-jian, SUN Lu-shi. Mechanism of Hg removal by gaseous advanced oxidation process with Fe3O4 and H2O2[J]. J Chem Ind Eng (China),
2018, 69(5):
1840-1845.
[29]
XU Z M, LV X J, CHEN J A, JIANG L X, LAI Y Q, LI J. First principles study of adsorption and oxidation mechanism of elemental mercury by HCl over MoS2(100) surface[J]. Chem Eng J,
2017, 308:
1225-1232.
doi: 10.1016/j.cej.2016.10.059
[30]
GUO P, GUO X, ZHENG C G. Computational insights into interactions between Hg species and α-Fe2O3 (001)[J]. Fuel,
2011, 90(5):
1840-1846.
doi: 10.1016/j.fuel.2010.11.007
[31]
HUANG D M, CAO D B, LI Y W, JIAO H J. Density function theory study of CO adsorption on Fe3O4(111) surface[J]. J Phys Chem B,
2006, 110(28):
13920-13295.
doi: 10.1021/jp0568273
[32]
KAUPP M, VON SCHNERING H G. Origin of the unique stability of condensed-phase Hg22+. An ab initio investigation of MI and MII species (M=Zn, Cd, Hg)[J]. Inorg Chem,
1994, 33(18):
4179-4185.
doi: 10.1021/ic00096a049
[33]
HU A, OTTO P, LADIK J. Relativistic all-electron molecular Hartree-Fock-Dirac-(Gaunt) calculations on HgO[J]. J Mol Struct,
1999, 468(3):
163-169.
doi: 10.1016/S0166-1280(98)00508-9
[34]
YANG Y, LIU J, ZHANG B, LIU F. Mechanistic studies of mercury adsorption and oxidation by oxygen over spinel-type MnFe2O4[J]. J Hazard Mater,
2017, 321:
154-161.
doi: 10.1016/j.jhazmat.2016.09.007
[35]
SU T, QIN Z, HUANG G, JI H, JIANG Y, CHEN J. Density functional theory study on the interaction of CO2 with Fe3O4(111) surface[J]. Appl Surf Sci,
2016, 378(15):
270-276.
[36]
ZHANG B, LIU J, YANG Y, CHANG M. Oxidation mechanism of elemental mercury by HCl over MnO2 catalyst:Insights from first principles[J]. Chem Eng J,
2015, 280:
354-362.
doi: 10.1016/j.cej.2015.06.056
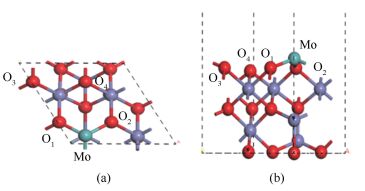
图 1
Mo/Fe3O4(111)Fetet结构图
Figure 1
Structure of Mo-doped Fe3O4(111) surface with Fetet termination
(a): side view; (b): front view(red: O atom; purple: Fe atom; green: Mo atom)

 下载:
下载:

 下载:
下载:







