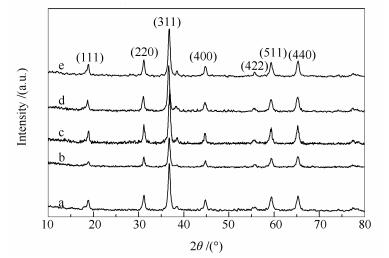
Figure 1.
XRD patterns of various Co3O4 catalysts synthesized by changing the CTAB concentrations
a: Co3O4(CTAB-0); b: Co3O4(CTAB-0.01); c: Co3O4(CTAB-0.03); d: Co3O4(CTAB-0.05); e: Co3O4(CTAB-0.06)

Nitrous oxide (N2O), as one of the greenhouse gases that were limited by the Kyoto protocol, has a global warming potential (GWP) of 310 and lifetime of 120 years in the atmosphere. In addition, N2O is also harmful to the ozone layer; if disposed to the atmosphere, it may cause great harm to our ecological environment. The production of nitric and adipic acid contributes mostly to the anthropogenic source of N2O. The decomposition of N2O to non-toxic and harmless N2 and O2 is a simple and practical method to reduce the N2O emission. Various catalysts were reported for the N2O decomposition, including supported noble metals[1-3], ion-exchanged zeolites[4-7], and transition metal oxides[8-10], among them, the Co3O4-based catalysts have been widely studied[11-16]. It was also observed in our previous work that the Co3O4 catalyst prepared by precipitation was highly active in the decomposition of N2O[17].
To enhance the performance of Co3O4-based catalysts in N2O decomposition, in this work, Co3O4 was hydrothermally synthesized by using hexadecyl trimethyl ammonium bromide (CTAB) as a template. It was reported that with the increase of CTAB concentration in aqueous solution, CTAB may be present in the form of monomers, pre-micelles, spherical, rod, and layered micelles; the first and second critical micelle concentrations (CMC) were 0.00089 and 0.021 mol/L, respectively, as measured by Liu et al[18]. If the CTAB concentration was higher than the second CMC, rod micelles were formed and cobaltosic oxide precursors were then produced. The Co3O4 catalysts were then prepared by calcining the cobaltosic oxide precursors, which was further modified with K by impregnation with K2CO3 solution and used in the decomposition of N2O under the atmosphere containing oxygen and steam. The effect of preparation parameters such as CTAB concentration, molar ratio of CTAB to cobalt, molar ratio of urea to cobalt, and K modification on the catalytic activity of Co3O4 was then investigated.
Appropriate amounts of Co(NO3)2·6H2O, urea, and CTAB was added into 50 mL deionized water; the mixture was stirred under ultrasonic irradiation for 10 min and then transferred to a 100 mL Teflon-sealed autoclave. The autoclave was then heated to 120 ℃ under rotation and held at this temperature for 6 h. The precipitant obtained was washed with deionized water, dried at 60 ℃ for 6 h, and then calcinated at 400 ℃ for 2 h. In this way, three series of cobaltosic oxides were prepared: the first one is denoted as Co3O4(CTAB-x), which is obtained with a CTAB/cobalt molar ratio of 1, a urea/cobalt molar ratio of 4, and a CTAB concentration of x (x = 0, 0.01, 0.03, 0.04, 0.05, and 0.06, in mol/L); the second one is expressed as Co3O4(CTAB/Co-y), prepared with 0.05 mol/L CTAB solution, a urea/cobalt molar ratio of 4, and a CTAB/cobalt molar ratio of y (y = 0.83, 1, 1.1, 1.3); the third one is expressed as Co3O4(urea/Co-z), synthesized with 0.05 mol/L CTAB solution, a CTAB/cobalt molar ratio of 1, and a urea/cobalt molar ratio of z (z = 3, 4, 5).
To prepare the K-modified Co3O4 catalyst with a K/Co molar ratio of 0.02 (0.02K/Co3O4), the Co3O4 catalyst prepared by using 0.05 mol/L CTAB solution, with a CTAB to cobalt molar ratio of 1 and a urea to cobalt molar ratio of 4 was then impregnated with K2CO3 solution at room temperature for 24 h, dried at 60 ℃ for 12 h, and then calcined at 400 ℃ for 2 h.
The decomposition of N2O was carried out in a fixed-bed reactor; 0.5 g catalyst was used for each test. Unless otherwise stated, the reactant feed consisted of 1% N2O and balanced argon, with a total flow rate of 150 mL/min. The effluent stream was analyzed by a gas chromatography (GC-920, Shanghai Haixin) equipped with a Porapak Q column and a thermal conductivity detector (TCD). In the case of oxygen atmosphere, the feed contained 2% O2 besides 1% N2O, whereas in the case of oxygen and steam atmosphere, the feed bore 8.2% H2O, besides 1% N2O and 2% O2.
The initial activity was expressed by the conversion of N2O measured after reaction at given temperature for 30 min. To test the catalytic stability of 0.02 K/Co3O4, the reaction was conducted at 400 ℃ for 50 h.
X-ray diffraction (XRD) patterns were recorded on a Shimadzu powder diffractometer (XRD-6100) with Cu Kα radiation and graphite monochromator at 40 kV and 30 mA.
Specific surface area was measured by nitrogen physisorption apparatus (NOVA3000, Quantachrome) and calculated by BET equation. Prior to the measurement, the catalyst samples were pre-treated at 300 ℃ for 2 h to remove any impurities; the nitrogen adsorption and desorption were performed at -196 ℃ and room temperature, respectively.
Scanning electron microscopy (SEM) images were taken on Hitachi S-4800; the catalyst samples were coated by using an ion sputter with platinum (E-1045, Hitachi) in order to improve their electric conductivity.
X-ray photoelectron spectra (XPS) of cobalt element were recorded in an ESCALAB250 spectrometer using Al Kα radiation with pass energy of 20 eV. The charging effect was corrected by referencing C 1s peak centered at 284.6 eV.
Temperature-programmed reduction by hydrogen (H2-TPR) was carried out on a chemical adsorption instrument (PCA-1200, Beijing Builder). Prior to each measurement, 80 mg catalyst was pre-treated in argon at 400 ℃ for 30 min and then cooled naturally to room temperature. Subsequently, the catalyst was exposed to 10% H2/Ar with a flow rate of 20 mL/min and heated from room temperature to 800 ℃ with a ramp of 10 ℃/min. The hydrogen consumption was detected by a TCD.
Temperature programmed desorption of oxygen (O2-TPD) were also performed on the PCA-1200 chemical adsorption apparatus. 100 mg catalyst was first exposed to O2 at 120 ℃ for 30 min; after cooling to ambient temperature, it was then heated in helium from room temperature to 700 ℃ at a ramp of 10 ℃/min, during which the desorbed oxygen was measured by the TCD.
A series of Co3O4 catalysts were first synthesized by changing the concentration of CTAB solution. As revealed by the XRD patterns shown in Figure 1, the Co3O4 catalysts have a spinel structure.
a: Co3O4(CTAB-0); b: Co3O4(CTAB-0.01); c: Co3O4(CTAB-0.03); d: Co3O4(CTAB-0.05); e: Co3O4(CTAB-0.06)
Table 1 illustrates that their BET surface area increases with an increase in CTAB concentration till 0.05 mol/L and then decreases with a further increase in CTAB concentration (e.g. Co3O4(CTAB-0.06)). The SEM images shown in Figure 2 suggest that Co3O4 prepared without using CTAB is in irregular particles. Irregular particles still appear with a low CTAB concentration (e.g. ≤ 0.01 mol/L). However, the Co3O4 catalysts prepared with 0.03-0.06 mol/L CTAB solution take the rod morphology.
 下载:
导出CSV
下载:
导出CSV
| Catalyst | BET surface area A/(m2·g-1) |
| Co3O4(CTAB-0) | 21.1 |
| Co3O4(CTAB-0.01) | 42.2 |
| Co3O4(CTAB-0.03) | 47.2 |
| Co3O4(CTAB-0.05) | 50.3 |
| Co3O4(CTAB-0.06) | 27.8 |

(a): Co3O4(CTAB-0); (b): Co3O4(CTAB-0.01); (c): Co3O4(CTAB-0.03); (d): Co3O4(CTAB-0.05); (e): Co3O4(CTAB-0.06)
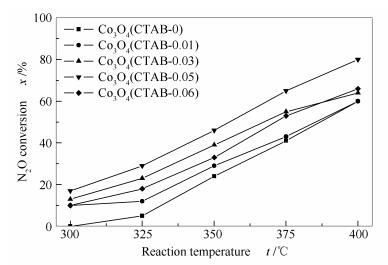
The conversion of N2O over various Co3O4 catalysts is displayed in Figure 3, which indicates that the Co3O4(CTAB-0.05) catalyst is most active in N2O decomposition. The reaction rate constants (k) can be derived by assuming the first order kinetics[19, 20]:
|
$ x = 1-\exp \left( {-k{V_{{\rm{bed}}}}/F} \right) $ |
(1) |
where x is the N2O conversion, Vbed is the catalyst bed volume, and F is the volumetric reactant flow rate.
The calculated rate coefficient (k), apparent activation energy (Ea) and pre-exponential factor (A) are then given in Table 2. Obviously, the Co3O4(CTAB-0.05) catalyst with higher activity has a lower apparent activation energy. Meanwhile, the values of Ea and A change in the same direction, that is, lnA = 3.53 + 0.159Ea, indicating that there is a dynamic compensation effect between Ea and A.
下载:
导出CSV
| Catalyst | k/s-1 | Ea/(kJ·mol-1) | lnA | ||||
| 300 ℃ | 325 ℃ | 350 ℃ | 375 ℃ | 400 ℃ | |||
| Co3O4(CTAB-0) | - | - | 1.96 | 3.77 | 6.55 | 84.2 | 16.9 |
| Co3O4(CTAB-0.01) | 0.75 | 0.91 | 2.45 | 4.02 | 6.55 | 74.5 | 15.2 |
| Co3O4(CTAB-0.03) | 1.01 | 1.87 | 3.53 | 5.70 | 7.30 | 65.4 | 13.8 |
| Co3O4(CTAB-0.05) | 1.33 | 2.45 | 4.40 | 7.50 | 11.50 | 69.8 | 14.9 |
| Co3O4(CTAB-0.06) | 0.75 | 1.42 | 2.86 | 5.39 | 7.71 | 77.1 | 15.9 |
Concerning the N2O decomposition mechanism, it was reported that N2O was first adsorbed on the catalyst surface and N-O bond was then broken; after that, two adsorbed oxygen atoms reacted to produce one oxygen molecule, which was then desorbed from the catalyst surface[21]. The reaction cycle over Co3O4 may follow the formula:
|
$ {{\rm{N}}_2}{\rm{O}} + {\rm{C}}{{\rm{o}}^{2 + }} \to {{\rm{N}}_2} + {\rm{C}}{{\rm{o}}^{3 + }}-{{\rm{O}}^-} $ |
(2) |
|
$ 2{\rm{C}}{{\rm{o}}^{3 + }}-{{\rm{O}}^-} \to {{\rm{O}}_2} + 2{\rm{C}}{{\rm{o}}^{2 + }} $ |
(3) |
where the desorption of oxygen species may be the controlling step.
Figure 4 shows the XPS spectra of various Co3O4 catalysts synthesized by changing the CTAB concentration. The binding energy of Co3+ 2p3/2 electrons on Co3O4(CTAB-0), Co3O4(CTAB-0.01), and Co3O4(CTAB-0.03) is about 782.2 eV, whereas that on Co3O4(CTAB-0.05) and Co3O4(CTAB-0.06) shifts about 0.1-0.2 eV to a lower value, indicating the Co3+-O- bond in Co3O4(CTAB-0.05) and Co3O4(CTAB-0.06) is weakened. As suggested by the second step of above mechanism, the weakened Co3+-O- bond in the (CTAB-0.05) and Co3O4(CTAB-0.06) catalysts is beneficial to the removal of oxygen species and the whole reaction cycle.
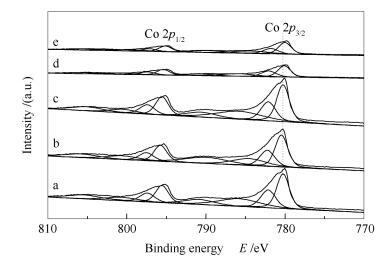
a: Co3O4(CTAB-0); b: Co3O4(CTAB-0.01); c: Co3O4(CTAB-0.03); d: Co3O4(CTAB-0.05); e: Co3O4(CTAB-0.06)
To extend the second step in above mechanism, it is assumed that two adsorbed oxygen atoms (Co3+-O-) produced by the dissocation of N2O molecules react with each other, forming one oxygen molecule O22- (i.e. Co3+-O-O-Co3+), which is then desorbed from the catalyst surface to regenerate the active Co2+ species:
|
$ 2{\rm{C}}{{\rm{o}}^{3 + }}-{{\rm{O}}^-} \to {\rm{C}}{{\rm{o}}^{3 + }}-{\rm{O}} - {\rm{O}} - {\rm{C}}{{\rm{o}}^3} \to {{\rm{O}}_2} + 2{\rm{C}}{{\rm{o}}^{2 + }} $ |
(4) |
O2-TPD is an effective method to characterize the interaction between oxygen species and catalyst surface. As shown in Figure 5, the oxygen desorbed in the O2-TPD profiles at 100-270 ℃ is assigned to the oxygen adsorbed on catalysts surface, whereas that desorbed at 330-420 ℃ is from the surface lattice oxygen. Obviously, the peak temperatures for oxygen desorption on Co3O4(CTAB-0.05) are 221.8 and 375.6 ℃, which are lower than those on other catalysts, indicating that the oxygen species on Co3O4(CTAB-0.05) are more easily desorbed, consistent with its higher activity.
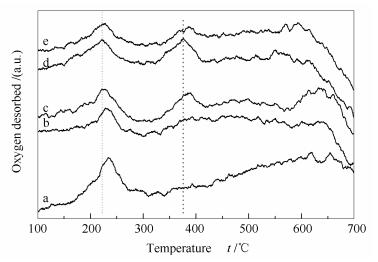
a: Co3O4(CTAB-0); b: Co3O4(CTAB-0.01); c: Co3O4(CTAB-0.03); d: Co3O4(CTAB-0.05); e: Co3O4(CTAB-0.06)
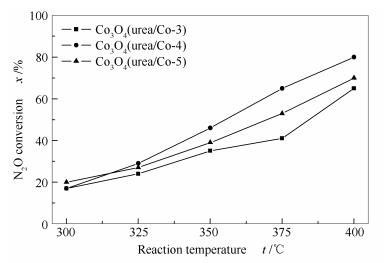
The effect of CTAB/cobalt and urea/cobalt molar ratios on the catalytic activity of Co3O4 was also considered. By changing the CTAB/cobalt molar ratio, it was found that the surface areas of Co3O4(CTAB/Co-0.83), Co3O4(CTAB/Co-1), Co3O4(CTAB/Co-1.1), and Co3O4(CTAB/Co-1.3) measured by nitrogen sorption are 40.3, 50.3, 38.8, and 36.7 m2/g, respectively. Meanwhile, by changing the urea/cobalt molar ratio, Co3O4(urea/Co-3), Co3O4(urea/Co-4), and Co3O4(urea/Co-5) have the surface areas of 42.1, 50.3, and 43.6 m2/g, respectively. The XRD results reveal that they all have the spinel structure.
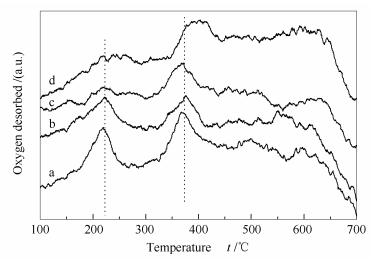
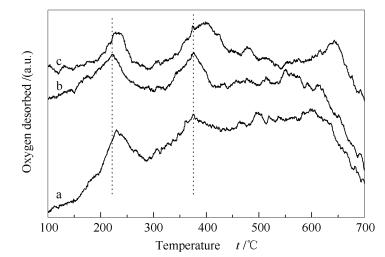
Figures 6 and 7 illustrate that the Co3O4 catalyst with a CTAB/Co molar ratio of 1 and a urea/Co molar ratio of 4 is most active, consistent with its high specific surface area. In the O2-TPD profiles shown in Figures 8 and 9.The oxygen desorbed at 100-270 ℃ is ascribed to the surface adsorbed oxygen, and that at 330-420 ℃ is assigned to the surface lattice oxygen. Obviously, the peak temperatures for the desorption of oxygen species on Co3O4 (CTAB/Co = 1 and urea/Co = 4) are 221.8 and 375.6 ℃, much lower than those on other catalysts.
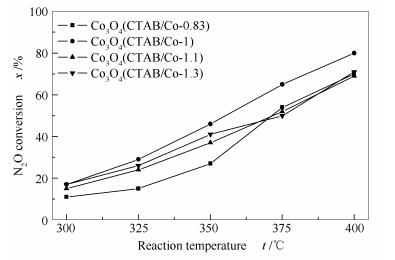
a: Co3O4(CTAB/Co-0.83); b: Co3O4(CTAB/Co-1); c: Co3O4(CTAB/Co-1.1); d: Co3O4(CTAB/Co-1.3)
a: Co3O4(urea/Co-3); b: Co3O4(urea/Co-4); c: Co3O4(urea/Co-5)
Above results illustrate that the Co3O4 catalyst synthesized by using 0.05 mol/L CTAB solution, with a CTAB/cobalt molar ratio of 1, and a urea/cobalt molar ratio of 4, exhibits high activity in N2O decomposition. To further enhance its catalytic activity, it was then modified by impregnation with K2CO3 solution. The K-modified 0.02 K/Co3O4 also shows the spinel structure and no crystallites of potassium species are observed. In comparison with K-free Co3O4, the surface area of 0.02 K/Co3O4 is decreased from 50.3 to 29.4 m2/g.
The cobalt XPS spectra shown in Figure 10 indicate that K-free Co3O4 has a Co2+/Co3+ molar ratio of 1.65, whereas K-modified 0.02 K/Co3O4 has a Co2+/Co3+ molar ratio of 1.69. For N2O decomposition, more active Co2+ species on 0.02 K/Co3O4 are beneficial to the adsorption and activation of N2O molecules. In addition, the binding energy of Co3+ 2p3/2 electrons on K-free Co3O4 is 782.1 eV, about 0.3 eV lower than that on 0.02 K/Co3O4, suggesting a weaker Co3+-O- bond on the K-modified 0.02 K/Co3O4 catalyst.
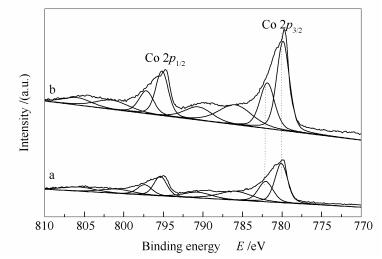
Figure 11 depicts the H2-TPR spectra of Co3O4 and 0.02K/Co3O4 catalysts.
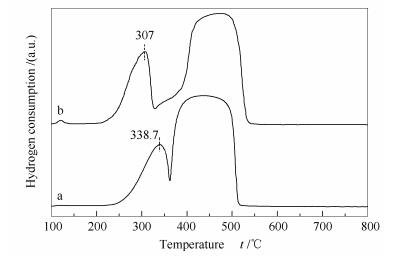
a: Co3O4; b: 0.02K/Co3O4
The low temperature peaks at 230-360 ℃ are ascribed to the reduction of Co3+ to Co2+, whereas the high temperature peaks at 360-540 ℃ are assigned to the reduction of Co2+ to metallic cobalt. For K-free Co3O4, the reduction of Co3+ to Co2+ appears at 338.7 ℃, whereas for K-modified 0.02 K/Co3O4, it is shifted to 307.0 ℃. It means that the Co3+-O- species on 0.02 K/Co3O4 is more easily reduced, consistent with the XPS result, which may contribute to the high activity of K-modified 0.02 K/Co3O4 (Figure 12).
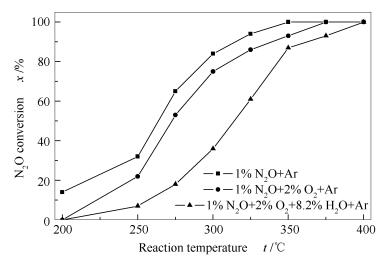
Figure 12 illustrates that when oxygen and steam are added into reaction feed, the conversion of N2O over 0.02 K/Co3O4 is decreased evidently. Table 3 gives the calculated rate coefficient (k), apparent activation energy (Ea) and pre-exponential factor (A). Obviously, oxygen and steam lead to a decrease in the reaction rate and an increase in the apparent activation energy; meanwhile, Ea and A values change in the same direction, viz., lnA = 6.95 + 0.144Ea, indicating that there is a dynamic compensation effect between Ea and A.
下载:
导出CSV
| Reaction feed | k/s-1 | Ea /(kJ·mol-1) | lnA | |||
| 250 ℃ | 275 ℃ | 300 ℃ | 325 ℃ | |||
| 1%N2O + Ar | 2.76 | 7.50 | 13.09 | 20.10 | 68.2 | 16.8 |
| 1%N2O + 2%O2 + Ar | 1.78 | 5.39 | 9.90 | 14.04 | 71.4 | 17.2 |
| 1%N2O + 2%O2 + 8.2%H2O + Ar | 0.52 | 1.42 | 3.19 | 6.73 | 88.5 | 19.7 |
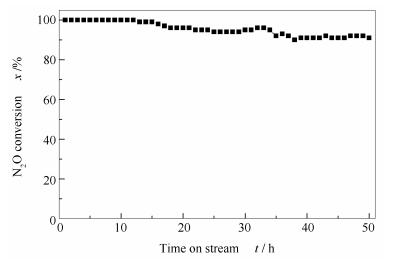
Figure 13 shows the catalytic stability of 0.02 K/Co3O4. Over the K-modified 0.02K/Co3O4 catalyst, the N2O conversion remains over 91% at 400 ℃ after conducting the reaction for 50 h in the presence of oxygen and steam, which is much more superior to the performance of Co3O4 prepared by precipitation reported in our previous work[17].
Cobaltosic oxide precursors were hydrothermally synthesized by using CTAB as the template and the Co3O4 catalysts were then prepared by calcining the cobaltosic oxide precursors, which was further modified by impregnation with K2CO3 solution and used in the decomposition of N2O; the effect of various preparation parameters on the catalytic activity of Co3O4 in N2O decomposition was then investigated.
The results indicated that the Co3O4 catalyst prepared by using 0.05 mol/L CTAB solution, with a CTAB to cobalt molar ratio of 1 and a urea to cobalt molar ratio of 4, exhibits high activity in N2O decomposition. The catalytic performance of Co3O4 can be further enhanced by modifying with K. Over the 0.02 K/Co3O4 catalyst, the N2O conversion remains over 91% at 400 ℃ after conducting the reaction for 50 h in the presence of oxygen and steam, which is much more superior to the performance of Co3O4 prepared by precipitation reported in our previous work.
LIN Y, MENG T, MA Z. Catalytic decomposition of N2O over RhOx supported on metal phosphates[J]. J Ind Eng Chem, 2015, 28: 138-146. doi: 10.1016/j.jiec.2015.02.009
PACHATOURIDOU E, PAPISTA E, DELIMITIS A, VASILIADES M A, EFSTATHIOU A M, AMIRIDIS M D, ALEXEEV O S, BLOOM D, MARNELLOS G E, KONSOLAKIS M, ILIOPOULOU E. N2O decomposition over ceria-promoted Ir/Al2O3 catalysts:The role of ceria[J]. Appl Catal B:Environ, 2016, 187: 259-268. doi: 10.1016/j.apcatb.2016.01.049
CARABINEIRO S A C, PAPISTA E, MARNELLOS G E, TAVARES P B, MALDONADO-HÓDAR F J, KONSOLAKIS M. Catalytic decomposition of N2O on inorganic oxides:Effect of doping with Au nanoparticles[J]. Mol Catal, 2017, 436: 78-89. doi: 10.1016/j.mcat.2017.04.009
XIE P F, MA Z, ZHOU H B, HUANG C Y, YUE Y H, SHEN W, XU H L, HUA W M, GAO Z. Catalytic decomposition of N2O over Cu-ZSM-11 catalysts[J]. Microporous Mesoporous Mater, 2014, 191: 112-117. doi: 10.1016/j.micromeso.2014.02.044
KUBONOVÁ L, PEIKERTOVÁ P, KUTLÁKOVÁ K M, JIRÁTOVÁ K, SŁOWIK G, OBALOVÁ L, COOL P. Catalytic activity of cobalt grafted on ordered mesoporous silicamaterials in N2O decomposition and CO oxidation[J]. Mol Catal, 2017, 437: 57-72. doi: 10.1016/j.mcat.2017.04.037
MELIÁN-CABRERA I, VAN ECK E R H, ESPINOSA S, SILES-QUESADA S, FALCO L, KENTGENS A P M, KAPTEIJN F, MOULIJN J A. Tail gas catalyzed N2O decomposition over Fe-beta zeolite. On the promoting role of framework connected AlO6 sites in the vicinity of Fe by controlled dealumination during exchange[J]. Appl Catal B:Environ, 2017, 203: 218-226. doi: 10.1016/j.apcatb.2016.10.019
SÁDOVSKÁ G, TABOR E, SAZAMA P, LHOTKA M, BERNAUER M, SOBALÍK Z. High temperature performance and stability of Fe-FER catalyst for N2O decomposition[J]. Catal Commun, 2017, 89: 133-137. doi: 10.1016/j.catcom.2016.10.029
MANIAK G, STELMACHOWSKI P, ZASADA F, PISKORZ W, KOTARBA A, SOJKA Z. Guidelines for optimization of catalytic activity of 3d transition metal oxide catalysts in N2O decomposition by potassium promotion[J]. Catal Today, 2011, 176: 369-372. doi: 10.1016/j.cattod.2010.11.043
LIU Z M, HE C X, CHEN B H, LIU H Y. CuO-CeO2 mixed oxide catalyst for the catalytic decomposition of N2O in the presence of oxygen[J]. Catal Today, 2017, 297: 78-83. doi: 10.1016/j.cattod.2017.05.074
LIU Z M, ZHOU Z Z, HE F, CHEN B H, ZHAO Y Y, XU Q. Catalytic decomposition of N2O over NiO-CeO2 mixed oxide catalyst[J]. Catal Today, 2017, 293/94: 56-60.
CHROMCÁKOVÁ Z, OBALOVÁ L, KOVANDA F, LEGUT D, TITOV A, RITZ M, FRIDRICHOVÁ D, MICHALIK S, KUSTROWSKI P, JIRÁTOVÁ K. Effect of precursor synthesis on catalytic activity of Co3O4 in N2O decomposition[J[[J]. Catal Today, 2015, 257: 18-25. doi: 10.1016/j.cattod.2015.03.030
OHNISHI C, ASANO K, IWAMOTO S, CHIKAMA K, INOUE M. Alkali-doped Co3O4 catalysts for direct decomposition of N2O in the presence of oxygen[J]. Catal Today, 2007, 120: 145-150. doi: 10.1016/j.cattod.2006.07.042
MANIAK G, STELMACHOWSKI P, KOTARBA A, SOJKA Z, RICO-PéREZ V, BUENO-LóPEZ A. Rationales for the selection of the best precursor for potassium doping of cobalt spinel based deN2O catalyst[J]. Appl Catal B:Environ, 2013, 136/137: 302-307. doi: 10.1016/j.apcatb.2013.01.068
GRZYBEK G, WÓJCIK S, CIURA K, GRYBO Ś J, INDYKA P, OSZAJCA M, STELMACHOWSKI P, WITKOWSKI S, INGER M, WILK M, KOTARBA A, SOJKA Z. Influence of preparation method on dispersion of cobalt spinel over alumina extrudates and the catalyst deN2O activity[J]. Appl Catal B:Environ, 2017, 210: 34-44. doi: 10.1016/j.apcatb.2017.03.053
YU H B, WANG X P, WU X X, CHEN Y. Promotion of Ag for Co3O4 catalyzing N2O decomposition under simulated real reaction conditions[J]. Chem Eng J, 2018, 334: 800-806. doi: 10.1016/j.cej.2017.10.079
WANG Y Z, HU X B, ZHENG K, WEI X H, ZHAO Y X. Effect of SnO2 on the structure and catalytic performance of Co3O4 for N2O decomposition[J]. Catal Commun, 2018, 111: 70-74. doi: 10.1016/j.catcom.2018.04.004
DOU Z, ZHANG H J, PAN Y F, XU X F. Catalytic decomposition of N2O over potassium-modified Cu-Co spinel oxides[J]. J Fuel Chem Technol, 2014, 42(2): 238-245. doi: 10.1016/S1872-5813(14)60016-5
LIU T Q, GUO R, SHEN M, YU W L. Determination of the diffusion coefficients of micelle and the first CMC and second CMC in SDS and CTAB solution[J]. Acta Phys Chim Sin, 1996, 12(4): 337-340.
STELMACHOWSKI P, MANIAK G, KOTARBA A, SOJKA Z. Strong electronic promotion of Co3O4 towards N2O decomposition by surface alkali dopants[J]. Catal Commun, 2009, 10(7): 1062-1065. doi: 10.1016/j.catcom.2008.12.057
PAN Y F, FENG M, CUI X, XU X F. Catalytic activity of alkali metal doped Cu-Al mixed oxides for N2O decomposition in the presence of oxygen[J]. J Fuel Chem Technol, 2012, 40(5): 601-607. doi: 10.1016/S1872-5813(12)60024-3
FRANKEN T, PALKOVITS R. Investigation of potassium doped mixed spinels CuxCo3-xO4 as catalysts for an efficient N2O decomposition in real reaction conditions[J]. Appl Catal B:Environ, 2015, 176-177: 298-305. doi: 10.1016/j.apcatb.2015.04.002

Figure 1 XRD patterns of various Co3O4 catalysts synthesized by changing the CTAB concentrations
a: Co3O4(CTAB-0); b: Co3O4(CTAB-0.01); c: Co3O4(CTAB-0.03); d: Co3O4(CTAB-0.05); e: Co3O4(CTAB-0.06)

Figure 2 SEM images of various Co3O4 catalysts synthesized by changing the CTAB concentration
(a): Co3O4(CTAB-0); (b): Co3O4(CTAB-0.01); (c): Co3O4(CTAB-0.03); (d): Co3O4(CTAB-0.05); (e): Co3O4(CTAB-0.06)

Figure 3 N2O conversions over various Co3O4 catalysts synthesized by changing the CTAB concentration

Figure 4 XPS spectra of various Co3O4 catalysts synthesized by changing the CTAB concentration
a: Co3O4(CTAB-0); b: Co3O4(CTAB-0.01); c: Co3O4(CTAB-0.03); d: Co3O4(CTAB-0.05); e: Co3O4(CTAB-0.06)

Figure 5 O2-TPD profiles of various Co3O4 catalysts synthesized by changing the CTAB concentration
a: Co3O4(CTAB-0); b: Co3O4(CTAB-0.01); c: Co3O4(CTAB-0.03); d: Co3O4(CTAB-0.05); e: Co3O4(CTAB-0.06)

Figure 6 N2O conversions over various Co3O4 catalysts synthesized by changing the CTAB/cobalt molar ratio

Figure 7 N2O conversions over various Co3O4 catalysts synthesized by changing urea/cobalt molar ratio

Figure 8 O2-TPD profiles of various Co3O4 catalysts synthesized by changing the CTAB/cobalt molar ratio
a: Co3O4(CTAB/Co-0.83); b: Co3O4(CTAB/Co-1); c: Co3O4(CTAB/Co-1.1); d: Co3O4(CTAB/Co-1.3)

Figure 9 O2-TPD profiles of various Co3O4 catalysts synthesized by changing urea/cobalt molar ratio
a: Co3O4(urea/Co-3); b: Co3O4(urea/Co-4); c: Co3O4(urea/Co-5)

Figure 10 Cobalt XPS spectra of the K-free and K-modified Co3O4 catalysts a: Co3O4; b: 0.02K/Co3O4

Figure 11 H2-TPR profiles of the K-free and K-modified Co3O4 catalysts
a: Co3O4; b: 0.02K/Co3O4

Figure 12 Activity of K-modified Co3O4 catalyst in N2O decomposition under different reaction atmospheres

Figure 13 Stability of the 0.02 K/Co3O4 catalyst in N2O decomposition at 400 ℃ under the atmosphere of 1%N2O, 2%O2, 8.2%H2O, and balanced argon
Table 1. BET surface area of various Co3O4 catalysts synthesized by changing the CTAB concentration
| Catalyst | BET surface area A/(m2·g-1) |
| Co3O4(CTAB-0) | 21.1 |
| Co3O4(CTAB-0.01) | 42.2 |
| Co3O4(CTAB-0.03) | 47.2 |
| Co3O4(CTAB-0.05) | 50.3 |
| Co3O4(CTAB-0.06) | 27.8 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Kinetic data for N2O decomposition over various Co3O4 catalysts synthesized by changing the CTAB concentration
| Catalyst | k/s-1 | Ea/(kJ·mol-1) | lnA | ||||
| 300 ℃ | 325 ℃ | 350 ℃ | 375 ℃ | 400 ℃ | |||
| Co3O4(CTAB-0) | - | - | 1.96 | 3.77 | 6.55 | 84.2 | 16.9 |
| Co3O4(CTAB-0.01) | 0.75 | 0.91 | 2.45 | 4.02 | 6.55 | 74.5 | 15.2 |
| Co3O4(CTAB-0.03) | 1.01 | 1.87 | 3.53 | 5.70 | 7.30 | 65.4 | 13.8 |
| Co3O4(CTAB-0.05) | 1.33 | 2.45 | 4.40 | 7.50 | 11.50 | 69.8 | 14.9 |
| Co3O4(CTAB-0.06) | 0.75 | 1.42 | 2.86 | 5.39 | 7.71 | 77.1 | 15.9 |
下载: 导出CSV
Table 3. Kinetic data of N2O decomposition over 0.02K/Co3O4 under different reaction atmospheres
| Reaction feed | k/s-1 | Ea /(kJ·mol-1) | lnA | |||
| 250 ℃ | 275 ℃ | 300 ℃ | 325 ℃ | |||
| 1%N2O + Ar | 2.76 | 7.50 | 13.09 | 20.10 | 68.2 | 16.8 |
| 1%N2O + 2%O2 + Ar | 1.78 | 5.39 | 9.90 | 14.04 | 71.4 | 17.2 |
| 1%N2O + 2%O2 + 8.2%H2O + Ar | 0.52 | 1.42 | 3.19 | 6.73 | 88.5 | 19.7 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们